Streiflichter Ausgabe 12/2021
Pharma Trends 2022
Jahrestagung des FORUM Instituts – „in Berlin und online erlebbar“
Nachberichterstattung durch Herrn Professor Sträter (zuerst publiziert als Streiflicht in Ausgabe 12/2021 der PharmInd).
Für den 22. und 23. November 2021 hatte das FORUM Institut zur traditionellen Jahrestagung „Pharma Trends 2022“ geladen, um einen Ausblick auf die Entwicklung Pharmamarktes im Jahre 2022 zu geben. Die Jahrestagung wurde als Hybridveranstaltung mit Teilnehmern präsent vor Ort in Berlin und anderen online im Büro oder Homeoffice durchgeführt. Bedingt durch die vierte Coronawelle hat sich kurz vor und im Verlauf der Tagung der Anteil der Onlineteilnehmer und der Onlinereferenten erhöht. Dies hat besondere Herausforderungen für die Konferenztechnik geschaffen, die aber sehr gut gemeistert wurden, so dass die Veranstaltung erfolgreich unter der Leitung von Frau Dr. Wolf-Klein und Herrn Prof. Sträter durchgeführt werden konnte.
Zu Beginn der Veranstaltung wurde bekannt, dass über das Wochenende das Ergebnis der Koalitionsverhandlungen für den Bereich Gesundheit und Pflege inoffiziell an die Presse gegeben wurde. Die Bundestagsabgeordneten der Ampel-Koalition haben daher ihre Teilnahme an einer Podiumsdiskussion absagen müssen, um der offiziellen Bekanntmachung der Koalitionsverhandlungen nicht vorzugreifen. Sie haben sich aber bereit erklärt, am 13. Dezember in einem Online-FORUM diese politischen Zielsetzungen zu kommentieren und zu erläutern. Nur der frühere CDU-Abgeordnete Hennrich wurde online zugeschaltet. Er zeigte sich nicht sonderlich überrascht. Im Bereich der Versorgung der Bevölkerung mit Generika seien keine Kostenersparnisse zu erzielen. Zusammenfassend wies er darauf hin, dass bei Koalitionsverhandlungen mit der CDU ein durchaus vergleichbares Ergebnis zu erwarten gewesen wäre!
Folgende Änderungen sind avisiert und wurden im Verlauf der Tagung diskutiert:
Die Versorgung mit Arzneimitteln soll durch Produktionsstandorte in der Europäischen Union gesichert werden. Das Nähere soll in gesetzlichen Regelungen bestimmt werden.
Der Zwangsrabatt für nicht festbetragsgeregelte Arzneimittel wird von 7 auf 16 % erhöht und gleichzeitig das Preismoratorium weiter fortgeschrieben. Das AMNOG-Verfahren wird weiterentwickelt. Der verhandelte Erstattungsbetrag wird auf den Beginn des 7. Monats nach Markteinführung rückwirkend angewendet und nicht erst auf den Beginn des 13. Monats. Die Mehrwertsteuer für alle Arzneimittel wird auf 7 % reduziert bei Fortschreibung des Preismoratoriums. Für die elektronische Patientenakte ePA soll das Prinzip des „opt out“ gelten, d. h. die Teilnahme bleibt freiwillig. Ein Ausschluss folgt allerdings erst dann, wenn der Patient ausdrücklich der Verwertung seiner Daten widerspricht.
Eines der großen Themen der Tagung war die „Sicherung der Arzneimittelversorgung in Europa“. Hierzu haben vorgetragen – aus gesundheitspolitischer Sicht – der Leiter der Abteilung 1 „Arzneimittel, Medizinprodukte und Biotechnologie“ im Bundesministerium für Gesundheit, Herr Ministerialdirektor Thomas Müller, und Frau Dagmar Wald-Eßer von IQVIA mit einer Analyse des deutschen und europäischen Marktes. Es wurde deutlich, dass die durch Corona ausgelösten Turbulenzen zu erheblichen Problemen in der Arzneimittelversorgung geführt haben. Die Politiker haben daher erkannt, dass „resiliente Lieferketten“ und Produktionsnetzwerke innerhalb der Europäischen Union wieder aufgebaut werden müssen. Die Beteiligten haben jedoch hervorgehoben, dass eine Anhebung der Generikapreise nicht automatisch zur Verlagerung der Produktionsstätten führt, sondern im Zweifel zu einer Erhöhung des Gewinns der Unternehmen, was nicht Ziel der politischen Anstrengungen sein könne. Ein Mittel ist die Ausschreibung von Rabattverträgen für Generika. Hier wird überlegt, ob für die Ausschreibung und Vergabe ein bestimmter Anteil der betroffenen Wirkstoffe und Arzneimittel aus europäischer Produktion gefordert werden muss. Wolfgang Späth, der Leiter der Abteilung Regulatory and external Affairs der Sandoz AG Deutschland hat die besondere Bedeutung von Biosimilars für die Versorgungssicherheit hervorgehoben. Ein großer Teil der Produktion findet sich unverändert in Europa. Die an medizinischen Fragen orientierte Substitution durch den verschreibenden Arzt hat sich nach Einschätzung der Generika/Unternehmen bewährt. Danach soll unbedingt eine aut-idem-Substitution auf der Grundlage von Rabattverträgen und durch Apotheker verhindert werden. Das Ziel der Reduktion von Ausgaben für biotechnologische Arzneimittel bei gleichzeitiger Wahrung der Versorgungssicherheit lasse sich auf diesem Wege am besten gewährleisten. Es wurde diskutiert, ob unter diesen Voraussetzungen noch eine Liste substituierbarer Biosimilars durch den Gemeinsamen Bundesausschuss zu rechtfertigen ist.
Zur Entwicklung der Ausgaben der gesetzlichen Krankenkassen für Arzneimittel von insgesamt ca. 43 Mrd. Euro pro Jahr tragen maßgeblich hochpreisige Arzneimittel bei. Die Preisbildungsmodelle für ATMPs waren daher ein wichtiges Thema. Dr. Dan Dammann, der Teamleiter Arzneimittel-Verordnungssteuerung der Techniker Krankenkasse – hat dazu ein viel diskutiertes „Fair Pricing Model“ vorgestellt, das von der International Association of Mutual Benefit Societies - AIM - entwickelt und in einem Modellversuch von der Techniker Krankenkasse und Prof. Gerd Glaeske am Beispiel verschiedener hochpreisiger Arzneimittel getestet wurde. Danach wäre eine Preisreduktion im Durchschnitt von ca. 30 % zu erzielen. Offen blieb jedoch die Frage, wer in welcher Verantwortung diese Preise festsetzen soll.
Die Abteilungsleiterin Arznei- und Heilmittel des GKV-Spitzenverbandes, Frau Dr. Antje Haas, hat ihre Vorstellung zur Reduktion der Ausgaben für hochpreisige Arzneimittel dargestellt. Sie gab insbesondere zu Bedenken, dass bei ATMPs die Annahme, dass nur eine Anwendung das Problem für den Patienten für immer löse, nicht erwiesen sei. Es lägen keine Langzeitdaten dafür vor, ob nicht später weitere Anwendungen notwendig werden könnten. Sie hat daher einen Interimspreis vorgeschlagen, der bis zur Gewährleistung des langfristigen Erfolges gezahlt werde, um dann später nach Garantie der langfristigen Wirksamkeit einen weiteren Ausgleich zu schaffen. Das von Herrn Dr. Dammann vorgestellte AIM Fair Pricing Model findet die ausdrückliche Zustimmung des GKV-SV.
Frau Dr. Haas wies darauf hin, dass ATMP-Arzneimittel für die Behandlung von Parkinson in der Entwicklung seien. Sollten diese erfolgreich sein, müsse neu nachgedacht werden, da diese Erkrankung sehr weit verbreitet sei und mit der Heilung über ATMPs zu einer Kostenexplosion führen könne.
Der Bewertung der Preisentwicklung haben sich auch angeschlossen Frau Daniela Teichert, die Vorsitzende des Vorstandes der AOK Nordost, und Prof. Dr. Christoph Straub, der Vorstandsvorsitzender der Barmer Krankenkasse. Sie bewerteten die bekannt gewordenen Koalitionsvereinbarungen der neuen Regierung sehr positiv und erläuterten anhand beeindruckender Zahlen die Herausforderung für die Finanzierung des GKV-Systems. So ist z. B. mit einer Zunahme der über 65-jährigen um 4 Millionen bis zum Jahre 2030 zu rechnen!
Frau Dr. Sylvia Demme, im Bundesamt für Soziale Sicherung verantwortliche Leiterin der Gruppe 31 „Risikostrukturausgleich - RSA“, hat sehr anschaulich dargestellt, wie hochpreisige Arzneimittel in der Gestaltung des morbiditätsorientierten Risikostrukturausgleichs berücksichtigt werden können. Es ist beeindruckend, wie eine kleine Abteilung von nicht einmal 50 Mitarbeitern im Bundesamt für Soziale Sicherung die Arzneimittelausgaben in Höhe von insgesamt ca. 245 Mrd. Euro in der GKV an über 100 Krankenkassen organisiert. Über 7 Mrd. Daten der GKV-Versorgung pro Jahr werden mithilfe von Algorithmen daraufhin ausgewertet, ob und in welcher Höhe Krankenkassen fair an den Gesamteinnahmen beteiligt werden. Von besonderer Bedeutung ist die Berücksichtigung von hochpreisigen Therapien im RSA. Es wurde ein Risikopool geschaffen, der ca. 80 % der Leistungsausgaben der GKV für hochpreisige Arzneimittel in dem jeweiligen Jahr ausgleicht. Am Beispiel von Zolgensma bei spinaler Muskelatrophie wurde dies sehr anschaulich dargestellt. Beeindruckend war die Analyse der Auswirkungen von Pay-for-Performance (P4P) Modellen – für die Zahlungen aus dem Risikopool. Unternehmen und Krankenkassen verfolgen unterschiedliche Modelle z. B. Ratenzahlungsmodelle, in denen Therapiekosten in Raten durch die Krankenkassen an das Pharmaunternehmen ausgezahlt werden oder sog. Rückerstattungsmodelle „upfront“. Hier werden die Therapiekosten vollständig durch die Krankenkassen an das Pharmaunternehmen gezahlt. Bei Therapieversagen erfolgt eine Rückerstattung an die Krankenkassen. Letzteres kann zu Verwerfungen führen, weil die Rückerstattung nach der aktuellen Gesetzeslage nicht an den Risikopool zurückgezahlt wird, sondern bei der Krankenkasse verbleibt, was für diese naturgemäß sehr attraktiv sein kann. Es sind jedoch bereits Strategien entwickelt, um solche Verwerfungen zu vermeiden.
Ein weiteres wichtiges Thema war die neue Verordnung für die Anwendung neuer Gesundheitstechnologien, die sog. EU Health Technology Assessment – HTA-Verordnung. Die Schlussabstimmung über den endgültigen Text im Plenum des Europäischen Parlaments soll im Dezember 2021 erfolgen. Deutschland hat im Trilogverfahren im Europäischen Rat bereits zugestimmt. Es bedarf allerdings noch der Zustimmung durch den Bundestag, die aber als gesichert gilt. Herr Thomas Müller, der Leiter der Abteilung Arzneimittel des BMG und die Leiterin der Abteilung Arzneimittel im Gemeinsamen Bundesausschuss, Frau Dr. Behring, haben die wesentlichen Prinzipien dieser neuen Verordnung dargestellt. Diese wird im nächsten Jahr in Kraft treten und erst drei Jahre später anwendbar sein, und zwar zunächst für Onkologika und ATMPs, drei Jahre später für Orphan Drugs, ca. 2030 für nononkologische Produkte und neue Medizinprodukte. In der Zwischenzeit laufen die Vorbereitungen auf die Umsetzung des neuen Systems „auf hohen Touren“.
In einem dem dezentralen Zulassungsverfahren vergleichbaren System soll eine Coordination Group mit Mitgliedern aus den Mitgliedstaaten gebildet werden, die eine Bewertung des Nutzens der Produkte vornehmen. Das Verfahren soll bereits parallel zum europäischen Zulassungsverfahren der EMA laufen. Gemeinsame wissenschaftliche Beratung, Scientific Advice und Scientific Consultation sollen organisiert werden, um das Anforderungsprofil für die betroffenen Unternehmen berechenbar zu machen. Spannend wird die Frage sein, ob und in welchem Umfang die gefundene Entscheidung der Coordination Group für die betroffenen Mitgliedstaaten verbindlich wird. Nach der HTA-Verordnung ist ein „taking into due concideration“ gefordert. Eine Ablehnung bedarf der Begründung. Sollte sich diese nicht überzeugen und häufiger vorkommen, ist, wie bei der Einführung des dezentralen Zulassungsverfahrens, mit Sanktionen der Europäischen Kommission zu rechnen. Entscheidend wird sein, ob das Verfahren die Beteiligung des G-BA ersetzen oder nur ergänzen wird. Hier war noch nicht ganz klar, welche Linie der G-BA verfolgen wird. Es ist allen Beteiligten zu wünschen, dass sich die Mitgliedstaaten nicht in „subtiler Obstruktion“ ergehen werden.
Besonders spannend war die Diskussionsrunde zwischen Aylin Tüzel, der Geschäftsführrin Pfizer Deutschland, Prof. Dr. Klaus Cichutek, dem Präsidenten des Paul-Ehrlich-Instituts sowie Dr. Ulrich Granzer, Granzer Regulatory Consulting & Services, und Dr. André Blümel, dem Vorsitzenden der PHAGRO, über die Erfahrungen in der Entwicklung, der Zulassung und dem Vertrieb von mRNA-Impfstoffen. Prof. Dr. Cichutek hat sehr anschaulich dargestellt, wie sich das Paul-Ehrlich-Institut schon sehr früh in den Prozess der Entwicklung durch Beratung der betroffenen Unternehmen eingeschaltet hat. Das Rolling Review Verfahren wurde positiv bewertet. Es erlaubt vor Antragstellung eine frühzeitige Bewertung von schon fertiggestellten Teilen des Dossiers und bewirkt damit eine Beschleunigung des Gesamtverfahrens. Es wurde deutlich, dass sich dies nicht ohne weiteres auf andere Zulassungsverfahren übertragen lässt, zumal die Erweiterung dieses Systems auf andere Arzneimittel zu Verzögerungen der verbleibenden Verfahren führen kann. Herr Dr. Granzer hat anschaulich die Herausforderung aus Sicht der Regulatory Affairs Abteilung dargestellt und die gute Kooperation zwischen Assessoren der Behörden und Unternehmen dargestellt. Eine besondere Herausforderung hat sich – von vielen unbemerkt –auch für die Distribution ergeben. Wenn der Bund kauft und bei der Bundeswehr einlagert, ist noch nicht gewährleistet, dass die Bundeswehr auch die Verteilung an die Apotheken oder Ärzte übernehmen kann. Hier ist vielmehr die Infrastruktur der vollversorgenden Großhändler gefordert. Herr Dr. André Blümel hat sehr beeindruckend dargestellt, wie gut hier die PHAGRO in Kooperation mit dem Bundesministerium für Gesundheit und der Bundeswehr eine effiziente Verteilung organisieren konnte. Vergleichbares ist zu erwarten bei der Versorgung mit Therapeutika zur Behandlung von COVID-19. Hier wurden die ersten zwei Arzneimittel - monoklonaler Antikörper - bereits zugelassen, und zwar für die Behandlung von stationär behandelten Patienten. Das neue Produkt von Pfizer Paxlovid läuft zurzeit im Rolling Review bei der EMA. Ein Zulassungsantrag ist – entgegen anderer Berichterstattung – noch nicht gestellt! Letzteres erscheint vielversprechend, weil es Substanzen aus der Gruppe der Protease Inhibitoren enthält, die auch zur Behandlung von AIDS erfolgreich eingesetzt werden. Die virusstatische Wirkung erlaubt es dem Patienten Antikörper aufzubauen, die dann auch einen langfristigen Effekt gewährleisten – im Unterschied zu HIV-Patienten, bei denen eine chronische Behandlung indiziert ist, weil nach Absetzen der Produkte die Viruslast wieder zunimmt. Vergleichbares ist in der Behandlung von COVID-19 nicht zu erwarten!
Der Präsident des BfArM, Prof. Karl Broich, hat das neue Forschungsdatenzentrum – FDZ – Gesundheit vorgestellt, das beim BfArM eingerichtet wird und Forschungseinrichtungen den Zugang erlauben soll. Unklar ist immer noch die Zugangsberechtigung von pharmazeutischen Unternehmen, obwohl beim Robert Koch Institut – RKI – eine Vertrauensstelle eingerichtet werden soll, die unabhängig vom BfArM eine Kontrolle insbesondere der datenschutzrechtlichen Aspekte durchführen kann. Es wurden die besonderen Herausforderungen in der Nutzung der Abrechnungsdaten deutlich, insbesondere ist eine Interoperabilität gefordert, die unter anderem durch die Verwendung einer einheitlichen Terminologie gewährleistet werden soll. Hier wird die Anwendung von Snomed CT in einem Pilotprojekt getestet. Hier eröffnen sich neue Perspektiven für die Analyse und Verwertung von Abrechnungsdaten aus dem GKV-System für die Entwicklung neuer Arzneimittel.
Über weitere Aspekte der Digitalisierung des Gesundheitswesens hat Dr. Markus Leyck Dieken, der Hauptgeschäftsführer der Gematik Berlin, vorgetragen. Er hat den Stand der Entwicklung der elektronischen Patientenakte – ePA - und des eRezepts auf der Grundlage der neuen Regelungen im 11. Kapitel des SGB V dargestellt. Die Patienten sollen danach ihre ePA-Daten ab 2023 freiwillig und pseudonymisiert für die Forschung freigeben. In der Diskussion ist ein „opt out“ System, d. h. solange der Patient nicht ausdrücklich widerspricht, ist eine Verwertung möglich. Die Umsetzung bereitet erhebliche Probleme, die Verhandlungen mit den kassenärztlichen Vereinigungen und dem Apothekerverband sind noch nicht abgeschlossen, so dass voraussichtlich eine Verlängerung der Übergangsfrist über den 01.01.2022 hinaus erforderlich wird. Erwähnung fand auch die Übersicht von Prof. Sträter zur „Digitalisierung der Arzneimittelversorgung in der GKV nach dem Digitale-Versorgung-Gesetz (DGV) und dem Patientendaten-Schutz-Gesetz (PDSG)“ im Streiflicht Pharm. Ind. 82, Nr. 8, 951-954 (2020).
Großes Interesse hat auch der Vortrag von Nico Reinhold zur digitalen Kommunikation in Healthcare Unternehmen gefunden. Er hat sehr anschaulich dargestellt, wie in sozialen Medien das Interesse der Verbraucher geweckt und bedient werden kann. Interessant war dabei, dass die sachliche Information über Nutzen und Risiko von Arzneimitteln in Schriftform mehr Vertrauen findet als in Videos.
Zur jüngsten Entwicklung im AMNOG-Verfahren hat vorgetragen Frau Dr. Behring, die Leiterin der Abteilung Arzneimittel im Gemeinsamen Bundesausschuss (G-BA). Die in § 35a SGB V geschaffene Möglichkeit der Anordnung zur Durchführung von anwendungsbegleitender Datenerhebung – AbD – wurde eingehend erläutert. Es sind noch nicht sehr viele Präparate erfasst. Auf der Homepage des G-BA sind in der Rubrik Beschlüsse die betroffenen Präparate aufgeführt. Es handelt sich insbesondere um zwei Präparate zur Behandlung der spinalen Muskelatrophie und zur Behandlung der Myelofibrose. Es wurde deutlich, dass der G-BA orientiert an den Vorgaben des SGB V entscheiden wird. Daneben ist jedoch zu beachten, dass diese Studien klinische Prüfungen im Sinne des Arzneimittelgesetzes sein können und daher zusätzlich der Genehmigung durch die zuständigen Bundesoberbehörden entweder das Paul-Ehrlich-Institut oder das BfArMs bedürfen. Auch die Beteiligung von Ethikkommissionen ist erforderlich. Das Bundesministerium für Gesundheit hat in seinem Schreiben für die Änderung der Verfahrensordnung des G-BA klargestellt, dass randomisierte Studien mit schwerwiegenden Interventionen nicht mehr als AbDs gewertet werden können.
Auch der Bereich der Reserveantibiotika und ihre Privilegierung im AMNOG-Verfahren wurde diskutiert. Hier wurde ersten Präparaten die Privilegierung im AMNOG-Verfahren gewährt. Einzelne Unternehmen gehen aber auch den Weg der Orphan Arzneimittel, der abhängig vom jeweiligen Produkt verschiedene Vorteile gegenüber der Qualifikation als Reserveantibiotikum bietet.
Zu den Erfahrungen und Problemen der Schiedsstelle im AMNOG-Verfahren hat deren Leiter, Prof. Dr. Stefan Huster vorgetragen. Er ist Inhaber des Lehrstuhls für Öffentliches Recht, Sozial- und Gesundheitsrecht und Rechtsphilosophie an der Ruhr-Universität Bochum und Leiter der Schiedsstelle nach § 130b SGB V. Er hat sehr anschaulich dargestellt, dass die vom Gesetzgeber durch unbestimmte Rechtsbegriffe geschaffenen Regelungslücken von der Schiedsstelle und der Rechtsprechung geschlossen werden müssen. Wenn im Gesetz vorgesehen ist, dass Arzneimittel mit belegtem Zusatznutzen einen „Zuschlag“ auf die Kosten der zVT erhalten sollen, so stellt sich die Frage, ob die Vervielfachung der Gesamtkosten der zVT noch als „Zuschlag“ qualifiziert werden kann. Die Schiedsstelle begrüßt ausdrücklich, wenn die Unternehmen und der GKV-SV in solchen Situationen zu vernünftigen Vergleichslösungen kommen. Er begrüßt ausdrücklich die Entscheidung des Bundesozialgerichts, mit der der sog. Albiglutid-Beschluss des LSG-BB vom 01.03.2017 aufgehoben worden ist (vgl. Streiflicht Pharm. Ind. 79, Nr. 5, Seite 602-603, 2017). Die hier geschaffenen vereinfachten Berechnungsmuster seien untauglich, innovative Forschung angemessen zu entlohnen.
Zu den jüngsten Entwicklungen in der Europäischen Union zur Gesetzgebung für Arzneimittel haben vorgetragen Dr. Alexander Natz von den Novacos Rechtsanwälten und Florian Schmidt, der stellvertretende Leiter der Einheit B5 in der Generaldirektion für Gesundheit und Lebensmittelsicherheit (DG Santé) der Europäischen Kommission. Schon Ende nächsten Jahres ist mit dem Entwurf einer Neuregelung für Orphan Arzneimittel und Kinderarzneimittel zu rechnen. Zurzeit werden die Erfahrungen der letzten ca. 20 Jahre ausgewertet und in einen Neuregelungsentwurf übertragen. Es ist damit zu rechnen, dass die Orphan Arzneimittel nach ihrer Prävalenz neu definiert werden und die Regelungen zu zur Intensivierung entsprechend modifiziert werden. Dabei kommt eine isolierte Änderung der Verordnung für Orphan Arzneimittel nicht in Betracht, da sie mit der Kinderverordnung interagiert. Es sollen daher gleich beide Verordnungen neu bearbeitet werden. In Diskussion ist auch, das gesamte Regelwerk der EU für Arzneimittel in den Richtlinien 2001/83/EU und 726/2004/EU in einen neuen einheitlichen Kodex zu überführen. Dieses Projekt ist jedoch unabhängig von der Überarbeitung der Regelung für Orphans und Kinderarzneimittel.
Zusammenfassend ist festzuhalten, dass unter der neuen Regierung der Arzneimittelmarkt in Deutschland nachhaltige Änderungen erfahren wird. Der Generikamarkt wird voraussichtlich geschont. Die Ausgaben für nicht festbetragsgeregelte Arzneimittel sollen beschränkt werden. Hier sind die Entwicklungen noch offen, aber die Zielsetzung erkennbar. Als vorläufige Maßnahme müssen die Erhöhung des Zwangsrabatts und die Rückwirkung des Erstattungsbetrages angesehen werden. Hier bedarf es keiner großen Maßnahmen zur Implementierung. Es sind schlicht die bereits vorhandenen und etablierten Systeme mit neuen Vorgaben auszustatten. Es bleibt abzuwarten, ob es bei dieser einfachen Geldschöpfung zum Auftakt bleibt oder auch andere nachhaltige Veränderungen in der Struktur zu erwarten sind.
Autor:
Rechtsanwalt Prof. Burkhard Sträter
Sträter Rechtsanwälte, Bonn
straeter@straeterlawyers.de
Bericht der Online-Weiterbildung „Verunreinigungen in Arzneimitteln – Fokus Nitrosamine“, FORUM Institut, 12. November 2021
Nitrosamine in Arzneimitteln: relevant in Entwicklung, Zulassung, Qualitätskontrolle und Überwachung
von Dr. Sabine Paris, GMP-Verlag Peither AG
„Verunreinigungen in Arzneimitteln – Fokus Nitrosamine“ titelte eine Online-Weiterbildung vom FORUM Institut am 12. November 2021. Vier ausgewiesene Fachleute aus Behörde, Industrie und Beratung beleuchteten alle Aspekte zu Nitrosaminen, die im Lebenszyklus eines Arzneimittels eine Rolle spielen: Regulatorische Anforderungen, Entwicklung, Verantwortung des Herstellers und der Lieferanten sowie Analytik und GMP-Überwachung.
Regulatorischer Rahmen
Dr. Andreas Grummel, Experte für pharmazeutische Qualität beim BfArM in Bonn, startete in den Seminartag mit einem Update zu den regulatorischen Anforderungen. Er erläuterte die wichtigsten Prinzipien der ICH Guideline M7(R1) on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk [1]. Mit der Einführung des Treshold of Toxicological Concern (TTC) wurde ein Paradigmenwechsel eingeläutet. Für genotoxische Substanzen, die potentiell in jeder Konzentration zu DNA-Schäden führen können, kann eigentlich keine sichere Dosis festgelegt werden. Aber für Arzneimittel wird ein Grenzwert benötigt! So wurde mit dem TTC-Prinzip eine als mit einem akzeptablen Risiko verbundene Dosis von 1,5 µg/Tag identifiziert. Der Wert ergab sich aus Extrapolationen von Daten aus der Carcinogenic Potency Database, wobei ein zusätzlicher Krebstodesfall auf 100.000 exponierten Personen als akzeptabel eingestuft wurde. Das ist ein rein formales und sehr konservatives Vorgehen. In den meisten Fällen wird das Risiko damit überschätzt. Dies gilt allerdings nicht für Substanzen des sogenannten „Cohort of Concern“, wie Aflatoxine, N-Nitroso und Azoxyverbindungen. Deren akzeptable Aufnahmemengen liegen meist weit unter dem TTC. Das potenteste Nitrosamin hat einen Acceptable Intake (AI) von 26,5 ng/Tag!
ICH M7 ist anwendbar für neue Wirkstoffe und neue Humanarzneimittel von der klinischen Entwicklung bis zur Zulassung. Zusätzlich greift die Guideline auch bei bereits zugelassenen Produkten, wenn
- Änderungen bei der Wirkstoffherstellung zu neuen Verunreinigungen führen oder bestehende Spezifikationen nicht mehr eingehalten werden können,
- Änderungen der Formulierung oder Produktzusammensetzung zu neuen Abbauprodukten führen oder bestehende Spezifikationen nicht mehr eingehalten werden können oder
- die Indikation oder Dosierung so geändert wird, dass sich das akzeptable Krebsrisiko für das Produkt verändert.
In einer Neubewertung werden alle Änderungen näher daraufhin betrachtet, ob sie das potentielle Risiko durch mutagene Verunreinigungen verändern können. Falls erforderlich,
sollte eine neue Prozesskontrollstrategie bei der Zulassungsbehörde eingereicht werden.
10 mögliche Ursachen für Nitrosamin-Verunreinigungen
Für Nitrosamin-Verunreinigungen führte Andreas Grummel folgende mögliche Ursachen an:
- Verwendung von Natriumnitrit oder anderen Nitriten in Gegenwart von sekundären oder tertiären Aminen oder quaternären Ammoniumsalzen
- Verwendung von Natriumnitrit oder anderen Nitrosierungsmitteln gemeinsam mit Reagenzien, Lösungsmitteln und Katalysatoren (Abbau zu sekundären oder tertiären Aminen)
- Verwendung von kontaminierten Rohstoffen im API-Herstellungsprozess
- Verwendung von rückgewonnenen Materialien (z. B. Lösungsmittel, Reagenzien, Katalysatoren)
- Kontaminierte Ausgangsmaterialien
- Kreuzkontaminationen
- Abbauprozesse von Ausgangsstoffen, Zwischenprodukten und APIs
- Verwendung bestimmter Verpackungsmaterialien, die Nitrocellulose enthalten
- Hilfsstoffe, die Nitrit enthalten
- Analytische Methoden, z. B. höhere Temperaturen bei Messungen können Nitrosaminbildung fördern
Nitrosamin-Verunreinigungen waren 2018 erstmalig in der Wirkstoffgruppe der Sartane festgestellt worden. Die Europäische Kommission hatte 2019 ein Referral-Verfahren nach § 31 der Richtlinie 2001/83/EG eingeleitet. Neben Bewertungen des Herstellungsprozesses wurde auch die Einführung einer Kontrollstrategie gefordert. Die ebenfalls überarbeiteten Ph. Eur.-Monographien legten Grenzwerte für N-Nitrosodimethylamin (NDMA) und N-Nitrosodiethylamin (NDEA) fest.
Nach dem Ende der Übergangszeit von 2 Jahren sind seit April 2021 0,03 ppm als Grenzwert für beide Nitrosamine einzuhalten. Dieser Wert ergibt sich aus dem technischen Limit des analytischen Verfahrens. Das kumulative Risiko der Nitrosamine für eine Krebserkrankung darf bei lebenslanger Einnahme nicht größer sein als 1:100.000.
Auf Antrag des Direktors der EMA nach Artikel 5 Absatz 3 der Verordnung (EG) Nr. 726/2004 hat der Ausschuss für Humanarzneimittel (CHMP) Leitlinien zur Vermeidung von Nitrosamin-Verunreinigungen in Humanarzneimitteln erarbeitet [2].
„Ein Artikel 5(3)-Verfahren ist rechtlich nicht bindend“, so die persönliche Meinung des Referenten. In Deutschland hätten bislang auch nur ca. 80 % der Zulassungsinhaber diesbezügliche Unterlagen eingereicht.
Überprüfung in drei Schritten
Die Leitlinien des CHMP sehen eine Überprüfung in drei Schritten vor. Im ersten Schritt wird eine Risikobewertung durchgeführt, die ICH Q9 Qualitätsrisikomanagement und ICH M7 berücksichtigt. Wenn in Schritt 1 ein Risiko der Nitrosaminbildung festgestellt wurde, muss im zweiten Schritt ein Bestätigungstest durchgeführt werden unter Verwendung von validierten und ausreichend empfindlichen Methoden. Die Ergebnisse der Bestätigungstests sollten spätestens bis zum 26. September 2022 an die Zulassungsbehörde übersandt werden. Allerdings sollten Zulassungsinhaber (MAHs) die zuständigen Behörden unverzüglich informieren, wenn die Tests das Vorhandensein von Nitrosaminen im Arzneimittel bestätigen.
Das BfArM hat eine analytische Methode (LC-MS-MS) entwickelt, mit der 13 verschiedene Nitrosamine in einem Analysengang identifiziert werden können. Die diesbezügliche Publikation wird bald erwartet.
Nach dem Stichtag im September 2022 ist das Thema aber noch nicht erledigt: Die Risikobewertung ist jeweils neu durchzuführen, wenn neue potentielle Ursachen für Nitrosamine identifiziert werden.
Für NDMA und NDEA sind für Sartane mit Tetrazolring akzeptable Grenzwerte festgelegt worden. Vorläufige Grenzwerte für andere Nitrosamine können auf Basis der maximalen Tagesdosis und der Behandlung über die gesamte Lebensdauer berechnet werden. Als „Default-Limit“, wenn aufgrund von fehlenden Daten kein Wert bestimmt werden kann, ist – sehr konservativ – 18 ng/Tag festgelegt worden.
Skip Testing gemäß ICH Q6A ist zulässig bei Werten ? 30 % des AI-Grenzwertes. Skip Testing erlaubt Prüfungen bei der Freigabe von vorausgewählten Chargen und/oder in vorher festgelegten Intervallen. Bei Werten konstant ? 10 % des AI ist sogar eine komplette Streichung des Tests aus der Spezifikation möglich. Die Ursache für die Verunreinigung muss jeweils bekannt sein.
Wenn mehrere Nitrosamine spezifiziert wurden, sind zwei Vorgehensweisen möglich, die jeweils zu begründen sind:
1. Die Summe aller Nitrosamine ist nicht größer als der AI vom potentesten Nitrosamin.
2. Das Gesamtrisiko der Nitrosamine ist nicht größer als 1:100.000.
In Schritt 3 des Verfahrens erfolgt dann die Änderung der Zulassungsdokumentation. Bis zum 26. September 2022 sollten die MAHs alle notwendigen Änderungen des Herstellungsprozesses des Wirkstoffs oder des Fertigarzneimittels beantragen.
Q&A-Papier der EMA
Das Q&A-Papier der EMA zu Nitrosaminen bezeichnete Andreas Grummel als gute Handreichung für die MAHs. Die einzelnen Fragen leiten durch den Prozess und geben Tipps z. B. zur Durchführung der Risikobewertung sowie der Bestätigungstests. Dieser Artikel greift einige der angesprochenen Fragen heraus.
„Die analytische Methode ist das A und O!“
Frage 9 des Q&A befasst sich mit den Analysemethoden, die ausreichend empfindlich sein müssen, um Spuren von Nitrosamin-Verunreinigungen nachweisen und quantifizieren zu können. In Schritt 2 ist daher auch die Bestimmungsgrenze (LoQ) zu zeigen. Wird eine quantitative Bestimmung durchgeführt, um auf eine Spezifikation verzichten zu können, sollte das LoQ ? 10 % des zulässigen Grenzwerts sein. Wenn mehrere Nitrosamine zu bestimmen sind, können auch unterschiedliche Methoden angewendet werden. Diskutiert werde im BfArM derzeit noch, ob es möglich ist, bei mehreren Nitrosaminen die Summe der einzelnen LoQs im Fertigarzneimittel zu verwenden, um zu beurteilen, ob 10 % des Risikos 1:100.000 unterschritten wird.
In Frage 10 werden Grenzwertberechnungen für Nitrosamine vorgestellt: Bestimmung des AI bei einer lebenslangen täglichen Verabreichung der maximalen Tagesdosis des Arzneimittels basierend auf ICH M7. Für einige spezifische Nitrosamine sind Grenzwerte festgelegt worden. Wenn es gar keine Daten geben sollte, kann der klassenspezifische TTC-Wert für Nitrosamine von 18 ng/Tag verwendet werden.
Frage 14 beleuchtet das Vorgehen bei neuen und laufenden Zulassungsanträgen. Eine Risikobewertung gemäß Schritt 1 sollte als Anlage zu Modul 1 mit einem Verweis auf Modul 3.2 eingereicht werden. Wird ein Risiko identifiziert, muss das Nutzen-Risiko-Verhältnis des Arzneimittels danach neu bewertet sowie eine Strategie zur Risikominimierung dargelegt werden. Auch sollten Pläne oder Daten zu Bestätigungstests gemäß Schritt 2 vorgelegt werden.
„Comply if tested: Eine hervorragende Lösung, gerade auch für die Sartane.“
Frage 15 eröffnet eine neue, interessante Möglichkeit zur Testung von Nitrosaminen. Wenn der Herstellungsprozess des Wirkstoffs als Ursprung der Nitrosamin-Verunreinigung identifiziert wird, können die Kontrolloptionen der ICH M7 genutzt werden, um nachzuweisen, dass die Verunreinigung im Fertigarzneimittel stets unterhalb des Grenzwertes liegt. Es muss dann ein Grenzwert in die Spezifikation des Fertigarzneimittels aufgenommen werden. Die getesteten Chargen sollten diesem entsprechen („Comply if tested“). Die Frequenz der Testung legt der MAH selbst fest. Anders ist das dagegen beim Skip Testing, wo eine definierte Frequenz bei der Behörde eingereicht werden muss.
Wichtige Links:
Lessons learned HMA/EMA: https://www.ema.europa.eu/en/documents/report/lessons-learnt-presence-n-nitrosamine-impurities-sartan-medicines_en.pdf
HMA/CMDh: Information on Nitrosamines for MAHs: https://www.hma.eu/620.html
Impurities – Strategien zur Kontrolle in Entwicklung, Produktion und Supply Chain
Dr. Michael Finkam, CMC Project Lead bei Grünenthal, stellte Hauptquellen von Verunreinigungen in Arzneimitteln vor, zeigte Strategien zur Kontrolle sowie ein mögliches Life Cycle Management von Verunreinigungen.
Quellen von Verunreinigungen sind:
- Synthese-Nebenprodukte
- Zersetzungsprodukte
- Reagenzien und Chemikalien
- Intermediate
- Metallrückstände
- Hilfsstoffe
- Wechselwirkung mit Verpackung oder Applikationsmaterialien
Die Wirkstoffherstellung ist eine der Hauptquellen. Die Kenntnis über Synthesenebenprodukte und Zersetzungsprodukte ist relevant für die Entwicklung der Analytik.
Während der präklinischen Entwicklung und den drei Phasen der klinischen Prüfung läuft das „Impurity Profiling“. Spezifikationen müssen für den Zulassungsantrag festgelegt werden. Aber z. B. in Phase III der klinischen Prüfung wird deutlich mehr Wirkstoff benötigt (mehrere 100 Kilo pro Jahr). Hier kommt es oft noch zu einem Wechsel des Herstellungsverfahrens und die Bewertung der möglichen Verunreinigungen muss neu aufgerollt werden. Zusätzlich wird die Formulierung der Arzneimittel im Laufe der Zeit noch optimiert. Dabei wird oft auch die Formulierungstechnologie gewechselt.
Die Verunreinigungen lassen sich in drei Klassen unterteilen: die tatsächlich aufgetretene Verunreinigung, die wissenschaftlich mögliche Verunreinigung und die hypothetische Verunreinigung (nicht wissenschaftlich naheliegend, aber theoretisch denkbar).
Als Monitoring-Tool eignet sich z. B. eine Übersichtsliste, die während der gesamten Produktentwicklung weitergeführt wird. Hilfreich ist auch ein „Impurity Gremium“, das interdisziplinär zusammengesetzt ist. Die technische Projektleitung übernimmt die Koordination. Teil des Gremiums sollten Qualitätssicherung/QP, analytische, chemische und pharmazeutische Entwicklung sowie die Toxikologie sein. Das Gremium legt das Verunreinigungsprofil fest sowie die Spezifikationslimits.
Die sieben Schritte des Impurity Assessments
Michael Finkam erläuterte die sieben Schritte des Impurity Assessments:
1. Schritt 1: Initiale Forced Degradation Studie
- Bestimmung der chemischen Stabilität
- Aufklärung und Herstellung der Hauptzersetzungsprodukte
Schritt 2: Liste der Verunreinigungen
- Reagenzien und Chemikalien
- Intermediate
- Synthese-Nebenprodukte
- Zersetzungsprodukte
•Schritt 3: Literatur und QSAR-Assessment (Die Datenbanken verknüpfen chemische Strukturen mit toxikologischen Befunden. So können mögliche Alert-Strukturen frühzeitig im Entwicklungsprozess gefunden werden.)
• Schritt 4: Klassifizierung der Verunreinigungen (ICH M7)
• Schritt 5: Bestätigung der positiven Strukturen
- AMES-Test (falls negativ, kann die Verunreinigung nach ICH Q3A/B behandelt werden)
• Schritt 6: Quantifizierung der identifizierten mutagenen Verunreinigungen
• Schritt 7: Umsetzung der Kontrollmaßnahmen (z. B. Grenzwerte auf der Stufe des Fertigarzneimittels festlegen)
Wann ist das Assessment zu wiederholen?
Anlässe für eine wiederholte Bewertung sind:
- Auftreten von neuen Verunreinigungen oder Zersetzungsprodukten
- Wechsel oder Anpassung der Syntheseroute
- Änderung der Formulierung
- Alle 2 Jahre Re-Assessment aller Strukturen in den QSAR-Datenbanken
„Der EFPIA-Guide zur Risikobewertung der Nitrosamine bildet den Stand der Wissenschaft sehr gut ab.“
Die Bewertung des Risikos für Nitrosamin-Verunreinigungen schließt die Suche nach Nitrosamin-Prekursoren (sekundäre und tertiäre Amine, nitrosierende Agenzien) sowie eine Kritikalitätsanalyse der einzelnen Herstellungsschritte ein. Michael Finkam empfahl den EFPIA Guide „Workflows for Quality risk management of nitrosamine risks in medicines“ [3], der wertvolle Hinweise und Hintergrundinformationen für die Bewertung und die Entwicklung einer Kontrollstrategie enthält. Insbesondere die ersten drei Guidances fassen die wichtigsten Erkenntnisse zu Quellen von nitrosierenden Agenzien, zu sekundären und tertiären Aminen sowie zu möglichen Kontaminierungsrisiken (z. B. wiederaufbereitete Materialien) zusammen. Für die Bewertung von Nitrosamin-Verunreinigungen in biologischen Arzneimitteln gibt die EFPIA ebenfalls Hinweise [4].
„Das Konzept der Forced Degradation ist Best Practice in der Pharmaindustrie.“
„Forced Degradation Studies“ erzwingen den Abbau eines Wirkstoffs durch drastische chemische und physikalische Einflüsse. Mit ihnen kann die Stabilität des Wirkstoffmoleküls untersucht und Abbauprodukte identifiziert werden. Wichtig ist, dass die Studien so gestaltet sind, dass sie realistische Ergebnisse liefern und keine Artefakte. Ein allgemeines Schema für die möglichen Stressbedingungen für den Wirkstoff und das Fertigarzneimittel zeigt Abbildung 1.
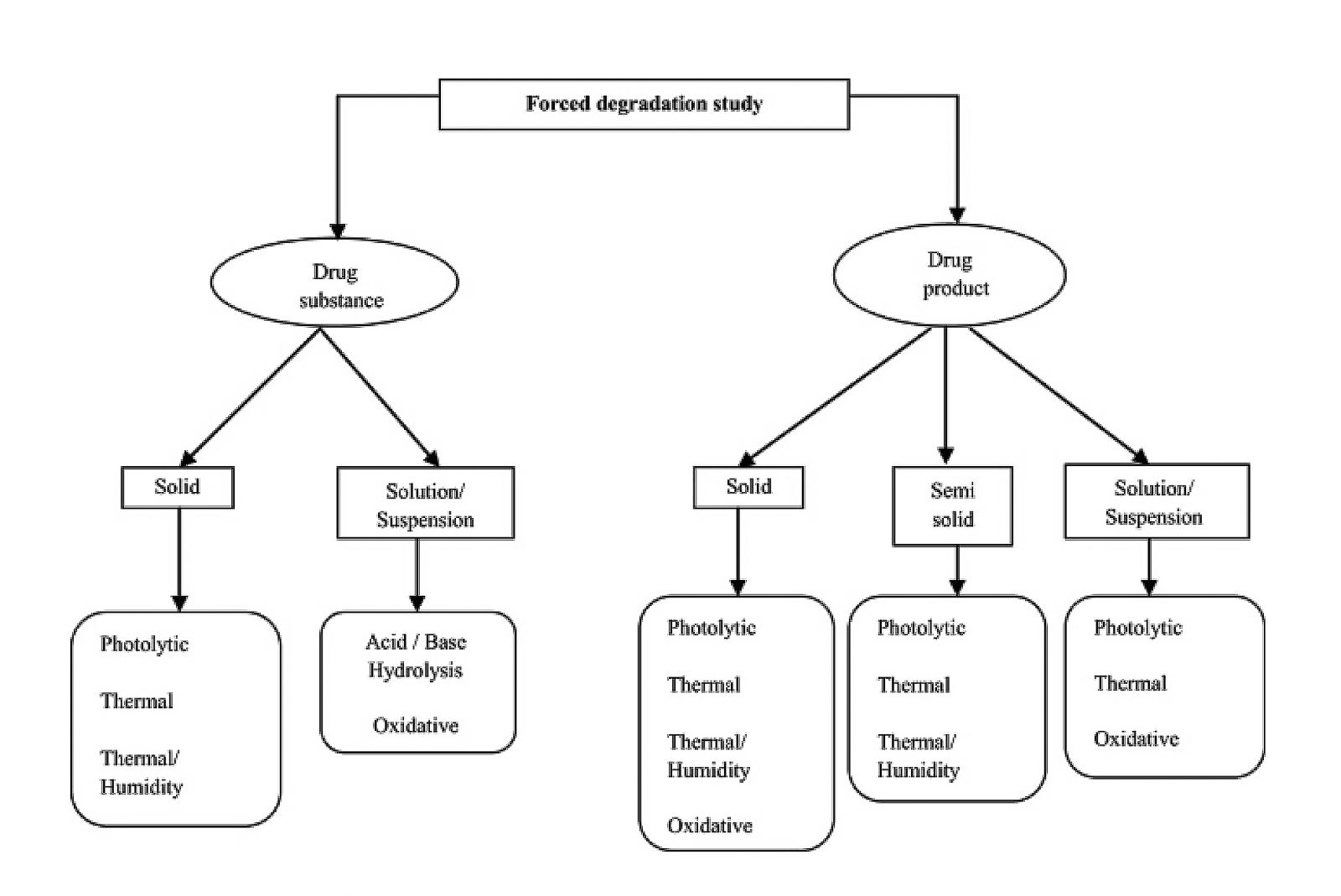
Abbildung 1
Unterschiedliche Stressbedingungen für die Zersetzung von Wirkstoff und Arzneimittel (Quelle: Blessy, M. et al.; J Pharm Analysis 2014 Jun;4(3):159-165)
Strukturaufklärung der Verunreinigungen
Herausfordernd für die Strukturaufklärung von Verunreinigungen ist, eine ausreichende Menge der Verunreinigung zu isolieren. Eine elegante analytische Methode koppelt eine HPLC (UV/MS) mit einer automatisierten Festphasenextraktion, die ein akkumulatives Trapping der Verunreinigungen ermöglicht, und einem NMR (NMR-SPE-LC).
Analytik von Verunreinigungen – Fokus Nitrosamine
„Die Analytik von Verunreinigungen ist (immer noch) eine Detektivarbeit“, führte Dr. Ralph Nussbaum, Auregen BioTherapeutics, in seinen Vortrag ein. Die analytischen Methoden sind unterschiedlich sensitiv. Im QC-Labor ist HPLC-MS die Standardmethode. Bei flüchtigen Verunreinigungen eignet sich die GC-MS-Kopplung. Zur Strukturaufklärung von unbekannten Verunreinigungen wird die NMR-Technik genutzt. Die hierfür notwendige Anreicherung der Verunreinigung erfolgt zumeist mit einer chromatographischen Trennsäule. Beim NMR werden heute auch insbesondere die heteronuklearen Verfahren HSQC (heteronuclear single quantum coherence) und HMBC (heteronuclear multiple bond correlation) genutzt.
„Wenn Sie nichts finden, heißt es nicht, dass nichts drin ist.“
2007 war der Proteaseinhibitor Nelfinavir-Mesylat (Viracept®) wegen einer Kontamination mit dem genotoxischen Ethylmethansulfonat (EMS) zurückgerufen worden. Die Verunreinigung stammte aus der Wirkstoffsynthese und betrug fast 0,1 %. EMS und Methylmethansulfonat (MMS) können mittels einer GC-MS-Methode identifiziert und quantifiziert werden.
2018 wurden mehrere Valsartan-haltige Arzneimittel zurückgerufen aufgrund einer Verunreinigung mit N-Nitrosodimethylamin (NDMA). NDMA gehört zu den potentesten karzinogenen Nitrosaminen. Zubereitungen müssen bereits ab 0,001 % gekennzeichnet werden. In untersuchten Stichproben der Arzneimittel wurden NDMA-Gehalte zwischen 3,7 und 22,0 Mikrogramm/Tablette gefunden. Ursache für die Verunreinigung war die Umstellung auf ein neues Syntheseverfahren, das Dimethylformamid (DMF) und Natriumnitrit nutzt. Diese reagieren nach Bildung des Nitrosylkations aus Natriumnitrit zu NDMA.
Aber auch andere Sartane enthielten NDMA, obwohl diese ganz anders synthetisiert werden. Ursache: Der Hersteller rezyklierte das Lösungsmittel DMF aus der Valsartan-Synthese und hat es für andere Synthesen eingesetzt.
Konsequenzen aus dem Valsartan-Fall
Aufgrund der Produktrückrufe kam es zu Lieferengpässen bei Sartan-haltigen Arzneimitteln. Jede Charge aller betroffenen Produkte musste auf Nitrosamine getestet werden. Die Analysenmethoden mussten produktspezifisch validiert werden. Das EDQM zog das für Valsartan erteilte CEP des betroffenen Herstellers zurück.
Die Ph. Eur.-Monographien der betroffenen Sartane wurden 2019 aktualisiert und Grenzwerte für NDMA und NDEA ergänzt.
Das EDQM hat verschiedene analytische Methoden entwickelt und publiziert. Überwiegend sind diese allerdings gekoppelte chromatographische Techniken, wie GC-MS und HPLC-MS, die oft nicht in QK-Routinelabors vorhanden sind.
Die niedrigen Grenzwerte der Nitrosamine sind analytisch herausfordernd.
Der EMA-Assessment Report zu dem Nitrosamin-Artikel 5(3)-Verfahren legt Grenzwerte für die acht häufigsten Nitrosamine fest und verweist auf gekoppelte Analysentechniken und Probenvorbereitung (z. B. Festphasenextraktion). Die sehr niedrigen Grenzwerte sind eine Herausforderung für die Analytik. Problematisch sind die Nachweisgrenzen. Die Grenzwerte, die Tagesdosis und die Flüchtigkeit der Nitrosamine bestimmen die Auswahl der Methode. Bei komplexen Matrices (wie Tabletten) kann mit der Stabilisotopen-Verdünnungsanalyse gearbeitet werden, die im MS die Empfindlichkeit erhöht und die Wiederfindung verbessert.
Dokumentation
Dr. Hiltrud Horn, HORN Pharmaceutical Consulting, beleuchtete, was Wirkstoffhersteller und Lieferanten von Hilfsstoffen berücksichtigen sollten, wie der Arzneimittelhersteller die Anforderungen effizient umsetzen kann und was für die USA relevant ist.
Grundsätzlich sollte bei Lieferanten sichergestellt sein, dass sie die neuesten Regelungen zu Nitrosaminen kennen und dass der Informationsfluss klappt. Ergebnisse früherer Inspektionen geben Hinweise auf die GMP-Compliance (z. B. via EudraGMDP-Datenbank oder U.S. FDA-483, Warning Letter). Eine regelmäßige Auditierung der Lieferanten ist sinnvoll.
Was ist zu beachten bei Wirkstoffherstellern?
Wirkstoffhersteller sollten dem MAH Informationen zu möglicher Nitrosaminbildung, zur Möglichkeit von Kreuzkontaminationen sowie eine Risikobewertung vorlegen können. Neben dem Herstellungsprozess selbst sind z. B. Kontaminationsquellen wie Ausgangsstoffe, Zwischenprodukte, Reagenzien und Lösungsmittel zu bewerten. Auch spielen Wasserqualitäten, das Vorhandensein von Stickstoffoxiden sowie die Primärverpackung eine Rolle.
In der Praxis unterstützen können die Fragebögen der APIC für Wirkstoffhersteller und die der IPEC für Hilfsstofflieferanten. Die EFPIA hat einen „Decision Tree“ für das „Drug Substance Manufacturing Process Risk Assessment for Presence of N-Nitrosamines“ veröffentlicht [5].
In der Risikobewertung sollte deutlich werden, welche Informationen zum Zeitpunkt der Erstellung vorlagen.
Wenn der Lieferant keine Informationen übersendet, kann der MAH in Schritt 1 der Nitrosaminbewertung angeben, dass ein Risiko für eine Nitrosamin-Verunreinigung besteht. Aber dann muss zwingend auch der Schritt 2 „Bestätigungstest“ durchgeführt werden. „Sobald Sie auf den Zug „Risiko“ gesprungen sind, fahren Sie weiter zur nächsten Station „Testen““, warnt Hiltrud Horn. Das etwaige spätere Einreichen von Informationen des Lieferanten ist dann nicht mehr ausreichend. Besser ist es, selbst nach wissenschaftlichen Erkenntnissen in der Literatur zu recherchieren (z. B. zum Syntheseweg) und auch andere Informationsquellen zu nutzen (Website, Auditbericht etc.).
Was ist zu beachten bei Lieferanten von Hilfsstoffen oder Packmitteln?
Nitrate und Nitrite sind in vielen Hilfsstoffen in Spuren enthalten. Riskante Hilfsstoffe sind z. B.:
- Natriumstärkeglykonat
- Croscarmellose-Natrium
- vorgelatinierte Stärke
- Polyvinylpyrrolidon (PVP), Kreuzpolyvinylpyrrolidon (cPVP)
- Laktose
NDMA/NDEA können auch während des Druckens von Deckelfolie gebildet werden. Die stickstoffhaltige Deckelfolie mit Nitrocellulose-Druckprimer reagiert mit Aminen in der Druckfarbe und erzeugt Nitrosamine. Diese werden während der Verblisterung (Heißversiegelung) auf das Produkt übertragen.
Wie kann der MAH die Anforderungen effizient umsetzen?
Der MAH ist verantwortlich für die Bewertung der potentiellen Nitrosamin-Verunreinigungen in seinen Arzneimitteln. Dabei ist das von der EMA geforderte dreistufige Verfahren anzuwenden (s. Abschnitt „regulatorisches Update“). Gegenüber den Behörden muss jeweils bestätigt werden, dass eine Risikobewertung (Schritt 1) bzw. Bestätigungstests (Schritt 2) durchgeführt worden sind. Die zugehörige Dokumentation wird nicht mitgesendet, sondern kann den Behörden auf Nachfrage vorgezeigt werden.
Was ist relevant für die USA?
Die Guidelines der U.S. FDA werden in der Regel zunächst als Entwurf zur Kommentierung veröffentlicht. Die Guidance „Control of Nitrosamine Impurities in Human Drugs“ dagegen wurde im September 2020 direkt in der finalen Version veröffentlicht und galt unmittelbar [6].
Im Gegensatz zu den europäischen Anforderungen gilt die FDA-Guidance nicht für biologische Arzneimittel. Auch differieren die zulässigen Grenzwerte für Nitrosamine. Reprocessing/Rework zur „Abreicherung“ der Nitrosamine ist gemäß FDA möglich. In der EU wäre dies schwer zu begründen und ist auch nicht Bestandteil des Zulassungsdossiers, so dass das Verfahren nicht registriert werden könnte.
Die Deadlines für die Einreichung der Nitrosamin-Bewertungen waren für Schritt 1 in der EU der 31. März 2021 und der 1. Juli 2021 (für biologische Arzneimittel), in den USA war es der 1. März 2021. Für die Übersendung etwaiger Änderungen der Zulassung (Schritt 3) sind die Deadlines der EU der 26. September 2022 und der 1. Juli 2023 (für biologische Arzneimittel) und die der USA der 1. September 2023.
Behördenaktivitäten zur Vermeidung von Nitrosaminen in Arzneimitteln
Dr. Franz Schönfeld, GMP-Inspektor bei der Regierung von Oberfranken, stellte die Aktivitäten und Erwartungen seiner bayerischen Überwachungsbehörde zum Thema Nitrosamine vor.
Er berichtete von dem allerersten „Nitrosamin-Fall“ im Juni 2018 in Deutschland, von dem ein in Nordbayern ansässiger Pharmazeutischer Unternehmer betroffen war. In ständigem Austausch mit dem BfArM und der EMA wurden europäische und internationale Maßnahmen abgesprochen und empfohlen (Sperrung der betroffenen Chargen des Fertigarzneimittels). Parallel liefen analytische Untersuchungen und toxikologische Bewertungen. Die Verunreinigung wurde als N-Nitrosodimethylamin (NDMA) identifiziert. Anfang Juli 2018 wurden europaweit alle Fertigarzneimittelchargen mit dem Wirkstoff Valsartan, der von Zhejiang Huahai Pharmaceuticals in China hergestellt worden war, zurückgerufen. Der chinesische Wirkstoffhersteller war in 160 Zulassungen als Hersteller angeführt.
„Die Kunst bei der chemischen Synthese ist die Isolierung des Wirkstoffs.“
NDMA entsteht bei der Synthese von Valsartan aus den zugegebenen Reagenzien DMF und Natriumnitrit bei sauren ph-Werten. Zur Zerstörung des überschüssigen Natriumazids (notwendig für die Synthese des Tetrazolrings) wird Natriumnitrit zugegeben („Quenching“). Das Nitrit reagiert mit sekundären Aminen (wie DMF) zu Nitrosaminen. Um das Valsartan möglichst Nitrosamin-frei zu isolieren, wird die unterschiedliche Polarität der einzelnen Inhaltsstoffe ausgenutzt. Nach Zugabe von Wasser und MTBE (Methyl-tert-butylether) entstehen zwei Phasen. Die unpolare Phase mit MTBE und Valsartan befindet sich oben. Die polare Phase mit DMF und Wasser ist unten. Hier ist es wichtig, dass auf eine saubere Phasentrennung geachtet wird (keine Emulsion!).
Die Überwachungsbehörden erwarten, dass der Arzneimittelhersteller eine strukturierte und fundierte Risikoidentifizierung und -analyse durchführt. Als Tools können Ursache-Wirkungs-Diagramme und Fehlerbaumanalysen genutzt werden. Eine Risikoanalyse kann z. B. mittels FMEA durchgeführt werden. Es geht darum, das anfängliche Risiko zu bewerten. Wie verändert sich die Bewertung, wenn risikominimierende Maßnahmen getroffen werden? Aber solange das mögliche Risiko nicht bestimmt ist, muss vom Worst Case ausgegangen werden.
Die Auftrittswahrscheinlichkeit einer Nitrosamin-Verunreinigung ist nur via Änderung des Herstellungsprozesses zu senken. Die Entdeckungswahrscheinlichkeit erhöht sich erst, wenn eine ausreichend empfindliche und validierte Analysenmethode entwickelt ist.
Gutes Mittel: Chargensperrung
Eine geeignete, vorläufige Maßnahme bei der Identifizierung eines möglichen Risikos ist eine Chargensperrung. Diese ist eine firmeninterne Maßnahme, die nicht nach außen dringt und zusätzlich reversibel ist. Die Sperrung kann wieder aufgehoben werden, falls die weiteren Daten und Bewertungen das Risiko nicht bestätigen.
Die Maßnahmen zur Risikokontrolle müssen mit den Behörden abgestimmt werden (über den Stufenplanbeauftragten). Die Risikokontrolle muss jeweils angepasst werden, wenn neue Daten/Erkenntnisse vorliegen.
Typische Fehler
„Die Lieferantenqualifizierung von Wirkstoffherstellern wird oft nur oberflächlich durchgeführt bzw. Auditberichte von schlechter Qualität eingekauft“, beklagt Franz Schönfeld. Der Arzneimittelhersteller ist aber verpflichtet, über detaillierte Informationen zum Herstellungsprozess des Wirkstoffs zu verfügen und seinen Wirkstoffhersteller zu qualifizieren und regelmäßig zu auditieren. Dies hat auch das EDQM im Jahr 2018 nochmals betont. In der Praxis kann eigentlich nur Wirkstoff von solchen Herstellern bezogen werden, die ihr Herstellungsverfahren gegenüber ihren Kunden offenlegen und einen guten Informationsaustausch pflegen.
Je mehr Arzneimittelhersteller sich die Wirkstoffsynthesen tatsächlich im Detail vor Ort ansehen, desto besser werden die Herstellungsverfahren.
Auch die Berichte zur analytischen Methodenvalidierung sollte sich der Arzneimittelhersteller näher ansehen. Nachweis- und Bestimmungsgrenze können falsch berechnet worden sein (z. B. falsche Bereiche beim Signal-Rausch-Verhältnis gewählt, um geschönte Zahlen zu bekommen).
Die Methode kann auf dem Papier validiert sein, aber z. B. die Wiederfindungsrate ist trotzdem nicht bestimmt worden. Diese ist aber relevant, da eine zu niedrige Wiederfindungsrate auch falsch niedrige Nitrosaminwerte vortäuschen kann. Ein Wechsel des Extraktionsmittels kann z. B. 25 % ausmachen bei der Wiederfindung und somit auch beim Analysenergebnis.
Zusammenfassung:
Nitrosamine sind ein facettenreiches Thema, das alle Pharmazeutischen Unternehmer nicht nur heute, sondern auch in Zukunft weiter begleiten wird.
Nitrosamin-Verunreinigungen in Arzneimitteln können verschiedene Ursachen haben. Am häufigsten werden Nitrosamine über die Wirkstoffsynthese eingebracht. Mögliche Ursachen sind aber auch z. B. rückgewonnene Lösungsmittel, Abbauprodukte oder Verpackungsmaterialien mit Nitrocellulose.
Die Leitlinien des Ausschusses für Humanarzneimittel (CHMP) sehen für alle (Human-)Fertigarzneimittel ein dreistufiges Bewertungsverfahren vor. Im ersten Schritt wird eine Risikobewertung durchgeführt. Wenn in Schritt 1 ein Risiko der Nitrosaminbildung festgestellt wurde, muss im zweiten Schritt ein Bestätigungstest durchgeführt werden unter Verwendung von validierten und ausreichend empfindlichen Methoden. In Schritt 3 des Verfahrens erfolgt dann die Änderung der Zulassungsdokumentation.
Die Zulassungsinhaber finden in einigen Guidelines gute Hinweise für die Umsetzung in der Praxis sowie Fragebögen für Lieferanten, z. B. Q&A-Papier der EMA, EFPIA-Guide zur Risikobewertung, Fragebogen der APIC und der IPEC.
Das A und O sind Analysemethoden, die ausreichend empfindlich sein müssen, um Spuren von Nitrosamin-Verunreinigungen nachweisen und quantifizieren zu können.
Das EDQM hat verschiedene analytische Methoden entwickelt und publiziert. Überwiegend sind diese allerdings gekoppelte chromatographische Techniken, wie GC-MS und HPLC-MS, die oft nicht in QK-Routinelabors vorhanden sind.
Mit “Forced Degradation Studies” kann die Stabilität des Wirkstoffmoleküls untersucht und Abbauprodukte identifiziert werden. Herausfordernd für die Strukturaufklärung von Verunreinigungen ist, eine ausreichende Menge der Verunreinigung zu isolieren. Eine elegante analytische Methode koppelt eine HPLC (UV/MS) mit einer automatisierten Festphasenextraktion, die ein akkumulatives Trapping der Verunreinigungen ermöglicht, und einem NMR (NMR-SPE-LC).
Eine qualitativ hochwertige Lieferantenqualifizierung, inklusive Vor-Ort-Audits, ist wichtig, um das Risiko für Nitrosamin-Verunreinigungen zu minimieren. Dies hilft auch, die Herstellungsverfahren für Wirkstoffe zu verbessern.
Quellen:
[1] ICH M7(R1): https://www.ema.europa.eu/en/ich-m7-assessment-control-dna-reactive-mutagenic-impurities-pharmaceuticals-limit-potential
[2] Dokumente und Guidances der EMA zu Nitrosaminen: https://www.ema.europa.eu/en/human-regulatory/post-authorisation/referral-procedures/nitrosamine-impurities
[3]https://www.efpia.eu/media/580594/workflows-for-quality-risk-management-of-nitrosamine-risks-in-medicines.pdf
[4]https://www.efpia.eu/media/580595/n-nitrosamine-impurities-in-biological-medicinal-products.pdf
[5] APIC Template for report on the risk of potential presence of nitrosamine impurities: https://www.apic.cefic.org/publications.html
IPEC Questionnaire for Excipient Nitrosamines Risk Evaluation: https://www.ipec-europe.org/articles/questionnaire-for-excipient-nitrosamines-risk-evaluation.html
EFPIA Decision Tree: https://www.efpia.eu/media/580594/workflows-for-quality-risk-management-of-nitrosamine-risks-in-medicines.pdf
[6] https://www.fda.gov/regulatory-information/search-fda-guidance-documents/control-nitrosamine-impurities-human-drugs
Autorin
Dr. Sabine Paris
GMP-Verlag Peither AG