 Hier können Sie den gesamten Newsletter herunterladen.
Hier können Sie den gesamten Newsletter herunterladen.
Der MDR-Vorschlag zur Fristverlängerung - 'Kann der Schöpfer reparieren, was er schafft?'
Autor: Erik Vollebregt
(Originalartikel in Englisch)
Am 6. Januar 2023 war es endlich so weit: der Moment, auf den viele gewartet hatten - der MDR-Verlängerungsvorschlag wurde endlich veröffentlicht.
Er ist nicht in jeder Hinsicht so ausgefallen, wie ich es erwartet hatte. Erstens enthielt er auch eine Änderung der IVDR: eine Abschaffung der Verkaufsfrist, was diesen Vorschlag auch für die IVDR sehr relevant macht. Zweitens, aber logischerweise, enthält er auch eine Änderung für Sonderanfertigungen, die ich persönlich nicht erwartet hatte, die aber im Nachhinein sehr viel Sinn macht.
Der Vorschlag erinnerte mich an den epischen philosophischen Dialog in der Szene 'Ich will mehr Leben, Vater' im Film Blade Runner, in der der bald ablaufende Replikant Roy Tyrell, die Person, die ihn erfunden hat, trifft und für 'mehr Leben' plädiert, wobei er Tyrell die existenzielle Frage stellt: 'Kann der Erschaffer reparieren, was er macht?'
Tyrell sagt, dass er die Tatsachen des Lebens nicht ändern kann [man muss früher oder später zur MDR übergehen] und erklärt, wie schwierig es ist, Prozesse in einer Lebensform zu ändern, die bereits im Gange ist [wenn ein Gesetz einmal angenommen wurde, ist es immer problematisch und chaotisch, es zu ändern, und manchmal kann es sein, dass die bereits geschaffenen Probleme nicht behoben werden]. Es folgt eine intensive Diskussion über mögliche Wege, wobei Roy Vorschläge macht, die von Tyrell jeweils widerlegt werden. Tyrell beendet die Diskussion mit seinem Trost: 'Du wurdest so gut gemacht, wie wir dich machen konnten.', was der EU-Gesetzgeber zweifellos über die MDR denken wird. Hoffen wir, dass die MDR-Verlängerung nicht wie die Blade Runner-Szene endet, in der Tyrell die Konfrontation mit seiner wütenden Schöpfung, die sein Schicksal nicht akzeptiert, nicht überlebt.
Im Falle des MDR hat die Verlängerung ihren Preis, wie wir weiter unten sehen werden. Alte Weisheiten besagen, dass es so etwas wie eine kostenlose Verlängerung nicht gibt und dass man sich vor Leuten hüten sollte, die Geschenke mitbringen, wenn sie etwas von einem wollen. In diesem Fall treffen beide alten Weisheiten zu. Die MDCG warnt schon seit langem davor, dass alles, was an Überbrückungs- und Verlängerungsmaßnahmen unternommen wird, nur den Herstellern zugutekommen soll, die bereits Schritte zur Umstellung auf die MDR unternommen haben. Wenn Ihr Unternehmen auf Nummer sicher gegangen ist, werden Sie feststellen, dass Ihre Möglichkeiten im Rahmen dieses Vorschlags sehr begrenzt sind, und zwar absichtlich.
Lassen Sie uns diesen Vorschlag mit dem Wissen von heute auseinandernehmen. Der MDR-Teil des Vorschlags stellt Artikel 120 Abschnitte (2), (3) und (4) MDR vollständig auf den Kopf.
Änderungen in Artikel 120 (2): Gültigkeit von Zertifikaten
Es wird vorgeschlagen, Artikel 120 (2) MDR dahingehend zu ändern, dass die gültigen (AI)MDD-Zertifikate gültig bleiben oder (im Falle abgelaufener Zertifikate) ab dem Ablaufdatum auf dem Zertifikat für den Zeitraum bis zum risikoklassenbasierten Backstop-Datum im neuen Artikel 120 (3b) (31. Dezember 2027 oder 2028) wieder aufleben und gültig sind.
Wie bitte? Abgelaufene Zertifikate leben wieder auf?! Ja, aber nur unter den in dem neuen Artikel 120 (2) (a) und (b) genannten Bedingungen:
- Entweder haben Sie vor Ablauf der Bescheinigung für das Altprodukt oder ein 'Produkt, das dieses Produkt ersetzen soll', eine Vereinbarung mit einer benannten Stelle unterzeichnet ODER
- Sie verfügen über eine gültige Ausnahmeregelung gemäß Artikel 59 der MDR oder eine gültige Freistellung gemäß Artikel 97 der MDR (beides muss anscheinend nicht zum Zeitpunkt des Ablaufs der Bescheinigung in Kraft sein, was sinnvoll ist).
Eine wichtige Frage ist, was der Begriff 'ein Produkt, das als Ersatz für dieses Produkt bestimmt ist' bedeutet, denn dies ist sowohl für die benannte Stelle als auch für den Hersteller ein entscheidender Punkt. Dieser Begriff wird weder in den Erwägungsgründen noch in der Erläuterung des Vorschlags geklärt, ist aber für die Anwendung der geänderten Übergangsregelung von entscheidender Bedeutung. Muss dieses Produkt beispielsweise gleichwertig mit dem alten Produkt sein, das es ersetzen soll? Und wird dies dann anhand der Kriterien für die Gleichwertigkeit gemäß der MDR bewertet? Oder kann es sich um ein Produkt handeln, das einen breiteren Verwendungszweck hat? Niemand weiß das, und dieses Konzept muss von der Kommission oder der MDCG geklärt werden. Und wir alle wissen, was das bedeutet: Das wird so bald nicht geschehen.
Die Tatsache, dass diese Klausel die Gültigkeit bestehender und gültiger Altgerätezertifikate von Rechts wegen über das Ablaufdatum hinaus verlängert (auch wenn dieses Datum noch in der Zukunft liegt), macht aber auch - im Gegenzug - die Bedingungen von Artikel 120 (3b) - (3d) sofort auf das Gerät anwendbar (siehe Diskussion der Bedingungen weiter unten). Ob es Ihnen nun gefällt oder nicht: mit einem noch gültigen Zertifikat als Ergebnis der automatischen Anwendung von Artikel 120 (2) MDR werden Sie in das neue Regime gedrängt - Sie haben keine Wahl. Was bedeutet das für Sie?
Änderungen in Artikel 120 (3): gestapelte risikobasierte Neuheitsschonfristen
Interessanterweise folgt der MDR-Vorschlag zur Abwechslung einer früheren IVDR-Gesetzesänderung, die Anfang 2022 umgesetzt wurde und gestaffelte Neuheitsschonfristen nach Risikoklassen vorsah. Die MDR kopiert nun diesen Mechanismus, wie ich es als wahrscheinliche Option vorausgesagt habe, fügt aber zusätzliche Bedingungen für die Nutzung der verlängerten Neuheitsschonfristen hinzu, die darauf abzielen, die verlängerten Neuheitsschonfristen für Hersteller auszuschließen, die eine der Bedingungen von Artikel 120 (2) nicht erfüllen, damit die Zertifikate über ihr Ablaufdatum hinaus gültig sind:
- Entweder haben Sie vor Ablauf der Bescheinigung für das Altprodukt oder ein 'Produkt, das dieses Produkt ersetzen soll', eine Vereinbarung mit einer benannten Stelle unterzeichnet
ODER
- Sie verfügen über eine gültige Ausnahmeregelung gemäß Artikel 59 der MDR oder eine gültige Ausnahme gemäß Artikel 97 der MDR (beides muss zum Zeitpunkt des Ablaufs der Bescheinigung noch nicht in Kraft sein, was sinnvoll ist).
Schauen wir uns die Details der Änderungen in Artikel 120 (3) an. 'Aufgrund der Länge der Bestimmung wird Absatz 3 durch die Absätze 3a bis 3g ersetzt', heißt es in der Begründung.
Artikel 120 (3a) - Abweichung von Artikel 5 MDR
Das bedeutet, dass Sie, wenn Sie die Anforderungen des Vorschlags erfüllen (das Produkt fällt unter Artikel 120 (3b) oder (3c) MDR und Sie erfüllen die Bedingungen gemäß Artikel 120 (3d) MDR), von der Hauptanforderung der MDR befreit werden: dem Besitz einer gültigen CE-Kennzeichnung gemäß MDR.
120 (3b) - nähen oder nicht nähen?
Artikel 120 (3b) sieht eine Triage von Produkten vor, für die bereits eine (AI)MDD-Bescheinigung vorliegt. Die Einstufung erfolgt auf der Grundlage der Risikoklasse gemäß Anhang VIII der MDR (in der Vorschrift wird dies übrigens nicht ausdrücklich erwähnt, aber dies entspricht der Vorgehensweise im Korrigendum für die hochgestuften Produkte der Klasse I) und auf der Grundlage einer Qualifizierungsfrage, die viele für die Anforderungen an die bewährte Technologie (WET) gemäß Artikel 18 (3) MDR (Implantatkarte), Artikel 52 (4) (Konformitätsbewertung) und 61 (6) (b) MDR (Anforderungen an die klinische Bewertung) schmerzen wird: Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Platten, Drähte, Stifte, Clips und Verbinder. In der Erläuterung des Vorschlags werden sie ausdrücklich als WET bezeichnet. Dies bedeutet, dass komplexere Produkte mit diesen Bezeichnungen wahrscheinlich nicht in den Anwendungsbereich dieser definierten Gruppe fallen. In dieser Hinsicht kann das Team-NB-Positionspapier über einen risikobasierten Ansatz für Medizinprodukte, die von einer Implantatkarte ausgenommen sind, und Informationen, die dem Patienten mit einem implantierten Produkt gemäß Artikel 18.3 zur Verfügung gestellt werden müssen, ebenfalls für die Qualifizierung relevant sein, und für Zahnimplantate kann das Team-NB-Positionspapier über die Anwendbarkeit der Ausnahmeregelung auf enossale Zahnimplantate und Zahnimplantat-Abutments relevant sein.
Für die Triage-Logik siehe das nachstehende zusammenfassende Flussdiagramm. Bitte beachten Sie, dass dieses Flussdiagramm nicht den gesamten Prozess und alle zutreffenden Bedingungen beschreibt - es dient nur der Triage. Und es ist eine Verbesserung des Flussdiagramms, das ich ursprünglich über LinkedIn geteilt habe.
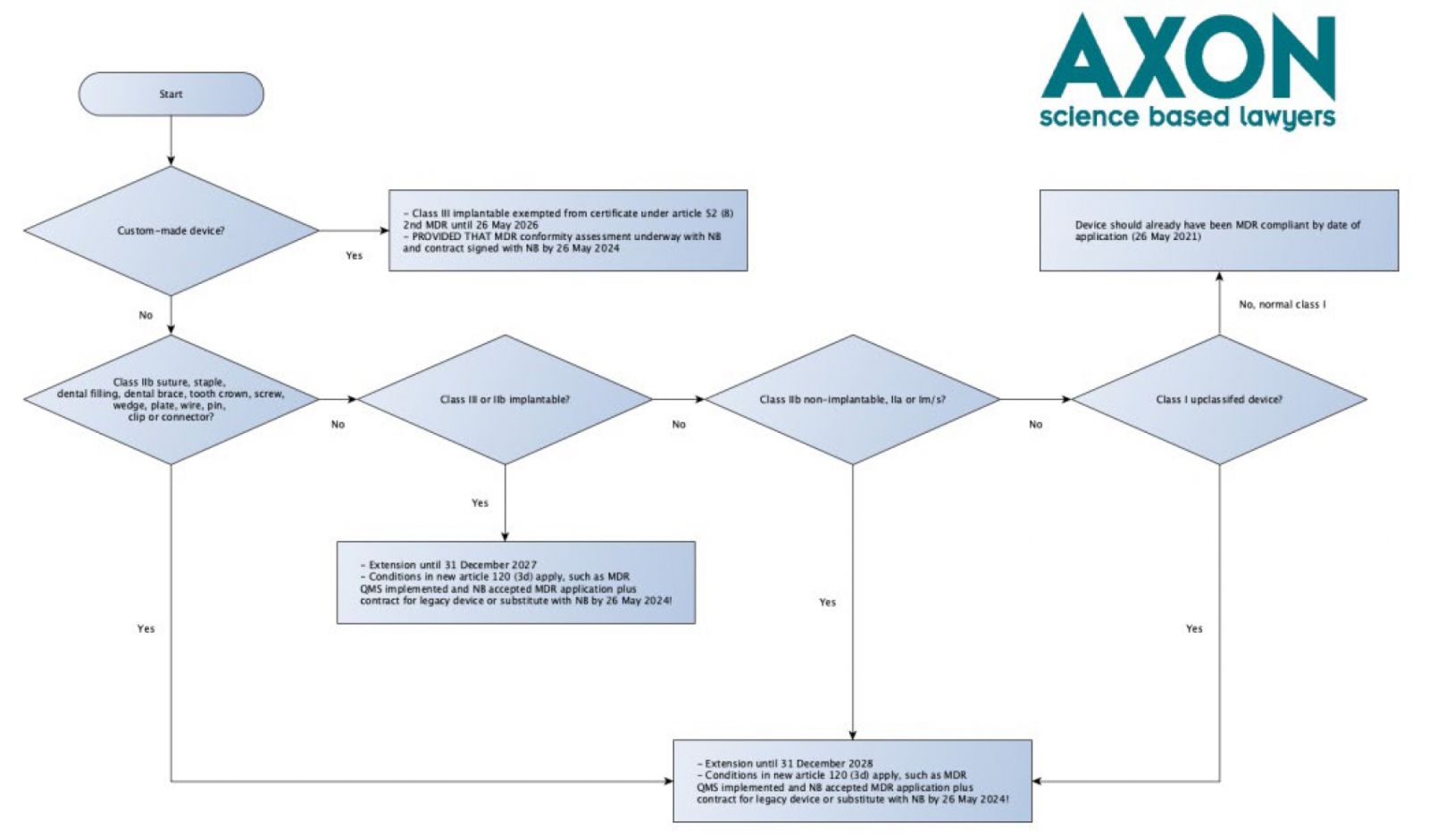
Triage-Flussdiagramm für MDR-Vorschlag
Ich hatte ursprünglich eine leicht abgewandelte Version des Flussdiagramms auf LinkedIn gepostet, in der Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Platten, Drähte, Stifte, Clips und Verbindungsstücke aufgrund der Verwendung des Kommas vor dem Wort 'außer' in Artikel 120 Absatz 3b (a) ('31. Dezember 2027, für Produkte der Klasse III und für implantierbare Produkte der Klasse IIb, ausgenommen Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Platten, Drähte, Stifte, Klammern und Verbindungsstücke') und die Begründung klarstellt
'Die Übergangsfrist wird vom 26. Mai 2024 bis zum 31. Dezember 2027 für Produkte mit höherem Risiko (implantierbare Produkte der Klasse III und der Klasse IIb mit Ausnahme bestimmter Produkte, für die die MDR Ausnahmen vorsieht, da diese Produkte als auf bewährten Technologien beruhend angesehen werden) und bis zum 31. Dezember 2028 für Produkte mit mittlerem und geringerem Risiko (andere Produkte der Klasse IIb und Produkte der Klassen IIa, Im, Is und Ir) verlängert.'
- Erläuternder Memorandumvorschlag
Später erfuhren wir, dass die Kommission in einer Antwort auf eine Frage klargestellt hatte, dass qualifiziertes Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Platten, Drähte, Stifte, Clips und Verbindungsstücke in den Geltungsbereich von 2028 fallen. Ich musste also mein ursprüngliches Flussdiagramm in die obige Version ändern. Verwenden Sie nicht die alte Version!
Was ich persönlich nicht sehr logisch finde, ist, warum die Fristenlogik von den 26. Mai-Fristen abweicht, die überall in der MDR und IVDR verwendet werden, und stattdessen den 31. Dezember 2027 und 2028 verwendet. Es ist natürlich schön, dass es mehr Zeit für den Übergang gibt, auch wenn es nicht logisch ist. Zumindest sind die Chancen, dass man damit in Schwierigkeiten gerät, gering, denn im schlimmsten Fall erfährt ein Unternehmen am 26. Mai, dass es bis zum 31. Dezember Zeit hatte.
Auch hier gibt es die Diskussion darüber, was ein 'Gerät, das dieses Gerät ersetzen soll' ist (siehe oben unter Artikel 120 (2) MDR).
120 (3c) - die höher eingestuften Produkte sollten sich mit der MDR beeilen
Artikel 120 (3c) befasst sich mit den höher eingestuften Produkten der Klasse I, oder anders ausgedrückt: mit den Produkten, die nach der MDD keine Bescheinigung der benannten Stelle benötigen, nach der MDR aber schon. Diese hatten den Korrigendum-2019-Bonus einer vor dem 26. Mai 2021 ausgestellten Konformitätserklärung erhalten, die bis zum Ende des Spielzeitraums gültig sein könnte, sofern die Anforderungen von Artikel 120 (3) MDR weiterhin erfüllt werden, wie z. B. keine wesentlichen Änderungen. Dies erwies sich insbesondere bei Software-Geräten als viel schwieriger als erwartet. Diese Geräte bekommen nun viereinhalb Jahre mehr Zeit: bis zum 31. Dezember 2028! Juhu!
Aber das hat seinen Preis: Für diese Geräte müssen Sie jetzt die Anforderungen gemäß Artikel 120 (3d) MDR erfüllen, zu denen nach wie vor gehören, dass bis zu diesem Datum keine wesentlichen Änderungen vorgenommen werden, und vor allem müssen Sie vor dem 26. Mai 2024 einige wichtige Dinge erledigen: Sie müssen ein vollständiges MDR-QMS implementiert haben (Artikel 120 (3d)) und einen Antrag auf Konformitätsbewertung bei einer benannten Stelle stellen, mit der Sie bis zu diesem Datum auch eine schriftliche Vereinbarung treffen müssen (Artikel 120 (3d) MDR). Das bedeutet, dass Sie als Hersteller eines höher eingestuften Produkts diese Bedingungen mehr als vier (4!) Jahre vor dem Ende Ihrer Übergangsfrist erfüllt haben müssen. Und wenn Sie das nicht tun, verlieren Sie die Gültigkeit Ihrer Konformitätserklärung bis zum 26. Mai 2024, und die MDD und MDR sind für Sie erledigt.
Denken Sie also kurz darüber nach: Sie werden wahrscheinlich nicht die volle Zeit bis Ende 2028 unter der MDD ausschöpfen können (es sei denn, Ihr Konformitätsbestätigungsverfahren dauert viereinhalb Jahre, was unwahrscheinlich ist). Wenn Sie also ein Gerät der Klasse I haben, haben Sie eigentlich keine Zeit zu verlieren! Sie haben weniger als anderthalb Jahre Zeit (bis zum 26. Mai 2024), um für die MDR-Konformitätsbewertung bereit zu sein - das ist doch etwas anderes, als bis zum 31. Dezember 2028 auf den Händen zu sitzen, oder? Ergo, machen Sie bitte nicht diesen Fehler. Die Zeit ist jetzt von entscheidender Bedeutung.
120 (3d) - es gibt keine kostenlose Verlängerung
So etwas wie eine kostenlose Verlängerung gibt es nicht, und auch diese ist an Bedingungen geknüpft. Diese Bedingungen sind in Artikel 120 (3d) festgelegt und - wohlgemerkt - sie gelten auch für Geräte der Klasse I, die, wie oben beschrieben, höher eingestuft wurden. Einige der Anforderungen sind bereits in der MDR enthalten (Artikel 120 (3) MDR): die Anforderung der kontinuierlichen Übereinstimmung mit der (AI)MDD (Artikel 120 (3d) (a) MDR) und keine wesentlichen Änderungen der Auslegung oder der Zweckbestimmung (Artikel 120 (3d) (b) MDR, wie in der MDCG 2020-3 klargestellt), was keine Änderung gegenüber der derzeitigen Situation darstellt. Bei den anderen drei Bedingungen in Artikel 120 (3d) ist dies jedoch mit Sicherheit der Fall, da sie neu sind (obwohl man dies für Artikel 120 (3d) (c) diskutieren könnte, der bereits Teil der angemessenen Überwachung für das (AI)MDD-Zertifikat ist).
Beginnen wir mit Artikel 120 (3d) (c), der verlangt, dass Ihr Produkt 'kein unannehmbares Risiko für die Gesundheit oder Sicherheit der Patienten, Anwender oder anderer Personen oder für andere Aspekte des Schutzes der öffentlichen Gesundheit darstellt'. Dies ist ein Begriff aus der Marktüberwachung (der in der MDR nur in den Marktüberwachungsbestimmungen in den Artikeln 93 bis 97 zu finden ist, was bedeutet, dass die zuständigen Behörden keine Schwierigkeiten mit dem betreffenden Produkt bekommen wollen). Aber wie wollen Sie das feststellen? Es ist Ihr Gerät, die (AI)MDD-Bescheinigung ist gültig, wie um alles in der Welt könnte es also ein inakzeptables Risiko darstellen? Der Grundgedanke einer gültigen (AI)MDD-Bescheinigung ist, dass das Gerät nur akzeptable Risiken aufweist. In der Erläuterung heißt es:
'Eine systematische Überprüfung der Sicherheit des Produkts ist nicht erforderlich, da Produkte, für die eine gemäß den Richtlinien ausgestellte Bescheinigung vorliegt, von der Stelle, die die Bescheinigung ausgestellt hat, oder einer gemäß der MDR benannten Stelle 'angemessen überwacht' werden. Stellt eine zuständige Behörde im Rahmen ihrer Marktüberwachungstätigkeit fest, dass ein Produkt ein unannehmbares Risiko für die Gesundheit oder Sicherheit von Patienten, Anwendern oder anderen Personen oder für andere Aspekte des Schutzes der öffentlichen Gesundheit darstellt, endet die Übergangsfrist für dieses Produkt.'
OK, und wie wird die benannte Stelle dies dann anwenden, da es sich nicht um eine Konformitätsnorm handelt, die einer angemessenen Überwachung unterliegt? Es sieht so aus, als beziehe sich dies auf das Kriterium in Abschnitt 4.5 der MDCG 2022-4 Rev. 1 ('wenn bei den Audittätigkeiten eine schwerwiegende Nichtkonformität festgestellt wird, die ein inakzeptables Risiko für die Gesundheit oder Sicherheit von Patienten, Anwendern oder anderen Personen darstellen kann'), das jedoch nach außen hin noch sehr unklar bleibt, aber die benennenden Behörden werden die benannten Stellen wahrscheinlich angewiesen haben, wie sie dies anzuwenden haben. Es wäre schön, wenn der Rest von uns das auch wüsste, oder? Das macht es so viel einfacher, die Anforderungen zu erfüllen.
In Artikel 120 (3d )(d) wird in der Erläuterung zum Vorschlag gefordert, dass
'der Hersteller bis spätestens 26. Mai 2024 ein Qualitätsmanagementsystem (QMS) gemäß Artikel 10 Absatz 9 der MDR eingerichtet hat. Mit dieser Bedingung soll sichergestellt werden, dass die Hersteller schrittweise zur vollständigen Einhaltung der MDR-Anforderungen übergehen. In diesem Stadium ist keine besondere Bescheinigung erforderlich, d. h. weder eine Selbsterklärung noch eine Überprüfung der Angemessenheit des QMS durch eine benannte Stelle. Durch die Einreichung eines Antrags auf Konformitätsbewertung bei einer benannten Stelle (siehe nächste Bedingung) bestätigt der Hersteller jedoch implizit, dass sein QMS den Anforderungen der MDR entspricht.'
-Erläuternder Memorandumvorschlag
Diese Bedingung zielt darauf ab, dass alle Hersteller von Altgeräten ab dem 26. Mai 2024 in Bezug auf das QMS gleichziehen, und ist ein Fortschritt gegenüber den Bedingungen, die bereits in Artikel 120 (3) MDR enthalten waren und die die Implementierung bestimmter Teile des QMS für Altgeräte vorschrieben. Dies wird nun auf das gesamte QMS-Konzept geändert.
Würde also die Anforderung, bis zu diesem Datum auch einen Antrag auf Konformitätsbewertung zu stellen, nicht eine Nichtanforderung aus dieser Bestimmung machen? Warum ist dies erforderlich, wenn die Bedingung nach 120 (3d) (e), dass ein Antrag auf Konformitätsbewertung für die MDR gestellt wurde, ausreicht? Das ist eine gute Frage. Denn das Problem mit gesetzlichen Anforderungen ist, dass man sie erfüllen muss, und das wäre strenger als eine implizite Bestätigung.
Was könnte also die Konsequenz sein? Das hätte - denke ich - nur dann Konsequenzen, wenn der Hersteller das QMS-Audit nach MDR nicht besteht (weil er dann kein konformes QMS eingerichtet hat), und die Folge der Nichterfüllung der Anforderung wäre die so genannte ex tunc-Ungültigkeit des Zertifikats, wobei sich herausstellt, dass der Hersteller die Anforderung nie erfüllt hat, am 26. Mai 2024 oder an dem Tag, an dem er den Antrag auf Konformitätsbewertung einreichte, nicht konform war, was bedeutet, dass die Bescheinigung ungültig war und der Hersteller nicht konforme Produkte auf den Markt gebracht hat, was im Rahmen der Marktüberwachung verboten ist und von den zuständigen Behörden mit Geldbußen, Rückrufaktionen usw. geahndet werden kann.
Schließlich verlangt Artikel 120 (3d) (e) als letzte Bedingung, dass der Hersteller oder ein Bevollmächtigter bis spätestens 26. Mai 2024 einen förmlichen Antrag gemäß Anhang VII Abschnitt 4.3 Unterabsatz 1 auf Konformitätsbewertung für ein in den Absätzen 3b und 3c dieses Artikels genanntes Produkt oder für ein Produkt, das als Ersatz für dieses Produkt bestimmt ist, gestellt hat und die benannte Stelle und der Hersteller bis spätestens 26. September 2024 eine schriftliche Vereinbarung gemäß Anhang VII Abschnitt 4.3 Unterabsatz 2 unterzeichnet haben. Beachten Sie, dass es sich dabei um das Altprodukt oder das 'Produkt, das als Ersatz für dieses Produkt bestimmt ist', handeln muss (siehe Artikel 120 (2) der MDR). Beachten Sie auch, dass diese Bedingung eine zusätzliche Bedingung im Vergleich zu Artikel 120 (2) (a) enthält: Es ist nicht nur eine schriftliche Vereinbarung erforderlich, sondern der Antrag auf Konformitätsbewertung muss bereits eingereicht worden sein.
Dies bedeutet, dass die Gültigkeit der Bescheinigung (die in Artikel 120 (2) (a) MDR behandelt wird) anders behandelt wird als das Inverkehrbringen/Inbetriebnahme unter dieser gültigen Bescheinigung (die in Artikel 120 (3d) behandelt wird): 'Geräte dürfen bis zu den in den Absätzen 3b und 3c dieses Artikels genannten Zeitpunkten nur in Verkehr gebracht oder in Betrieb genommen werden, wenn die folgenden Bedingungen erfüllt sind'). Ich bin mir nicht sicher, warum diese Entscheidung getroffen wurde. Sie lässt die Möglichkeit zu, eine gültige Bescheinigung zu haben (was für den Handel außerhalb der Union nützlich sein kann), aber nicht in der Lage zu sein, Geräte in der Union in Verkehr zu bringen. Es bedeutet jedoch auch, dass ein Hersteller vor dem 26. Mai 2024 einen Antrag auf Konformitätsbewertung stellen muss, um weiterhin Produkte in Verkehr bringen oder in Betrieb nehmen zu können. Auch hier stellt sich die Frage, was 'einen förmlichen Antrag gestellt' bedeutet: Muss der Hersteller einen Antrag gestellt haben oder muss der Antrag auch von der benannten Stelle validiert und akzeptiert werden? Letzteres geht weder aus dem Text noch aus dem Antrag hervor, so dass dies bedeuten kann, dass dies in der Praxis mit vielen Anträgen, die kurz vor dem 26. Mai 2024 gestellt werden, getestet wird.
Solange die Bescheinigung oder Konformitätserklärung gültig ist (also spätestens bis zum 26. Mai 2024), würden Sie davon ausgehen, dass die Bedingungen für die weitere Gültigkeit gemäß Artikel 120 Absätze 3b, 3c und 3d für Sie nicht gelten, und Sie könnten Ihre Bescheinigung in aller Ruhe bis zum Ablaufdatum aussitzen. Aber so funktioniert der Vorschlag nicht, wie oben beschrieben. Artikel 120 (2) in der geänderten Fassung verlängert diese Bescheinigungen von Rechts wegen unmittelbar nach Inkrafttreten des Vorschlags bis entweder zum 31. Dezember 2027 oder 2028, ohne Ihnen eine Wahlmöglichkeit einzuräumen. Artikel 120 (3d) (e) sieht vor, dass Sie einen Antrag auf Konformitätsbewertung bei einer benannten Stelle gestellt und eine Vereinbarung mit der benannten Stelle unterzeichnet haben müssen, damit die Bescheinigung oder die Konformitätserklärung gültig bleibt. Wie bereits erwähnt, bedeutet dies, dass Sie nicht warten können, sondern sofort mit der Vorbereitung und Einreichung der besten Konformitätsbewertung beginnen müssen.
Auch hier ist relevant, was passiert, wenn der Antrag abgelehnt wird: War das Inverkehrbringen und die Inbetriebnahme bis zu diesem Zeitpunkt rückwirkend rechtswidrig (ex tunc-Antrag) oder wird es nur für die Zukunft rechtswidrig sein (ex nunc-Antrag)? Der Vorschlag schweigt zu diesem Punkt.
120 (3e) - die alten Bedingungen des Artikels 120 (3) MDR bleiben bestehen
Die alten Bedingungen von Artikel 120 (3) bleiben für die alten Produkte bestehen (MDR Überwachung nach dem Inverkehrbringen, Marktüberwachung, Vigilanz, Registrierung der Wirtschaftsakteure und der Produkte), aber dies ist wirklich nur bis zum 26. Mai 2024 relevant, da der Hersteller nach diesem Datum ein vollständiges QMS nach Artikel 10 (9) MDR betreibt.
120 (3f) - Umstellung der benannten Stelle
Artikel 120 (3f) befasst sich mit der Frage, welche benannte Stelle für die Überwachung der Bescheinigung zuständig ist, wenn diese über ihr Ablaufdatum hinaus gültig bleibt (es muss sich dabei nicht um eine von der MDR bestimmte benannte Stelle handeln), und wie es funktioniert, wenn das Altprodukt durch ein anderes Produkt ersetzt wird. Die benannte Stelle der MDR, mit der die Vereinbarung gemäß Artikel 120 (3d) (e) unterzeichnet wird (diejenige, die für das Altprodukt oder das Ersatzprodukt vorhanden sein muss), übernimmt die Überwachung der benannten Stelle, die die Bescheinigung des Altprodukts oder des Ersatzprodukts überwacht hat, wenn diese benannte Stelle nicht gemäß der MDR benannt wurde.
120 (g) - Sonderanfertigungen haben die kürzeste Übergangszeit
Kundenspezifisch gefertigte Implantate der Klasse III (die gemäß Artikel 52 (8) der MDR eine Intervention der benannten Stelle erfordern) sind logischerweise auch in diesem Vorschlag enthalten.
Was ich nicht verstehe, ist, warum die Frist der 26. Mai 2026 ist und nicht der 31. Dezember 2026, und warum sie nicht in die Kategorie der Klasse III-Implantate mit dem 31. Dezember 2027 gegangen sind. Vielleicht ist die Kommission der Meinung, dass maßgefertigte Produkte einen einfacheren Weg haben oder so? Das ist nicht unbedingt der Fall.
Es gelten die gleichen Bedingungen wie in Artikel 120 (3d) (e): Unterzeichnung einer Vereinbarung mit einer benannten Stelle bis zum 26. September 2024 und Einreichung eines Antrags bis zum 26. Mai 2024.
Änderungen an Artikel 120 (4): Abschaffung der Verkaufsfrist
Mit den Änderungen an Artikel 120 (4) soll die Verkaufsfrist vollständig abgeschafft werden. Dies ist das Ergebnis einer erfolgreichen Lobbyarbeit, die gezeigt hat, dass die Verkaufsfrist einfach zu kurz war, weil die meisten Geräte lange brauchen, um die Lieferkette zu durchlaufen, und zwar langsamer als der Gesetzgeber ursprünglich angenommen hatte. Außerdem würden die Geräte, die in der Lieferkette stecken bleiben, bis zum 27. Mai 2025 für den EU-Markt verloren gehen, was ebenfalls eine schlechte Idee wäre. In den Worten des Vorschlags:
'Um zu verhindern, dass sichere Medizinprodukte und In-vitro-Diagnostika, die sich noch in der Lieferkette befinden, unnötigerweise entsorgt werden und damit die Gefahr einer drohenden Verknappung von Produkten erhöht wird, sollte die weitere Bereitstellung von Produkten zeitlich unbegrenzt sein.'
- Erwägungsgrund 10 des Vorschlags
Das bedeutet, dass der Verkauf von Altgeräten, die rechtmäßig in Verkehr gebracht wurden, zeitlich unbegrenzt sein wird und dass es einen beträchtlichen Zeitraum geben wird, in dem noch Altgeräte auf dem Markt sind, die verkauft werden und mit MDR-zertifizierten Geräten konkurrieren. Abgesehen von der Verwirrung, die zweifellos über den Konformitätsstatus von Altgeräten herrschen wird, die im Jahr 2030 immer noch an Endverbraucher verkauft werden (was durchaus möglich ist), stellt sich die Frage, wie fair dies gegenüber den Herstellern ist, die in die MDR-Umstellung investiert haben und möglicherweise für eine beträchtliche Zeit mit diesen Geräten konkurrieren müssen. Und für die Patienten, die möglicherweise mit alten Geräten behandelt werden, während modernere Geräte mit MDR-CE-Kennzeichnung verfügbar sind. Die Idee hinter der Ausverkaufsfrist war an sich nicht schlecht: dass zu einem bestimmten Zeitpunkt nur noch nach MDR-Standards zugelassene Geräte auf dem Markt sein würden.
Haushaltsführung (Artikel 122 und 123)
In Artikel 1 (2) und (3) des Vorschlags werden einige technische Anpassungen vorgenommen, indem die Artikel 122 (Aufhebung von Richtlinien) und 123 (verzögertes Inkrafttreten von Bestimmungen) geändert werden, um den Änderungen in Artikel 120 des Vorschlags Rechnung zu tragen.
IVDR-Änderungen: Abschaffung der Veräußerungsfrist
Für die IVDR sieht der Vorschlag die Abschaffung der Verkaufsfrist vor. Hintergrund ist, dass die Kommission erkannt hat, dass die Verkaufsfrist einfach zu kurz ist und dazu führt, dass die Produkte am Ende der Verkaufsfrist in der Lieferkette stecken bleiben. In den Worten von Erwägungsgrund 10 des Vorschlags:
'Um zu verhindern, dass sichere Medizinprodukte und In-vitro-Diagnostika, die sich noch in der Lieferkette befinden, unnötigerweise entsorgt werden und damit das Risiko eines drohenden Produktmangels erhöht wird, sollte eine solche weitere Bereitstellung von Produkten zeitlich unbegrenzt sein.'
- Erwägungsgrund 10 des Vorschlags
Ich muss sagen, dass ich dies nicht erwartet hatte. Dies mag aus den gleichen Gründen geschehen sein wie bei den MDR-Medizinprodukten, aber es wirft auch die gleichen Probleme auf wie bei den MDR-Medizinprodukten: Es wird Altprodukte auf dem Markt geben, die nach 'alten' Standards Zugang erhalten haben und mit nach IVDR-Standards bewerteten Produkten um einen potenziellen Marktanteil konkurrieren werden.
Bezug zur MDCG 2022-18 (Artikel 97 Ausnahmen)
Sollte der Vorschlag angenommen werden, wird die Ausdehnung von Legacy-Geräten nach geltendem Recht, die potenziellen Belastung der zuständigen Behörden durch die Inanspruchnahme von Artikel 97 der MDR, für den das MDCG 2022-18 die Vorlage war, massiv verringern.
Die Rolle der zuständigen Behörden wird sich darauf beschränken, sich mit den Ausnahmefällen zu befassen, die aus dem einen oder anderen Grund nicht in die Vorschlagslogik passen, so wie es ihnen gefällt und ihre übliche Arbeitsweise ist.
Bezug zum Positionspapier der MDCG 2022-14
Kein Vorschlag ist eine Insel. Wie ich in einem Interview mit RAPS festgestellt habe, brauchen wir auch das volle und unerschütterliche Engagement der MDCG und der Mitgliedsstaaten für die Umsetzung der MDCG 2022-14 Maßnahmen, um genügend Raum für die benannten Stellen zu schaffen, um der Herausforderung gerecht zu werden. Ich habe mich etwas kritisch über die Fähigkeit der MDCG geäußert, etwas zu leisten, aber ich möchte enthusiastisch bleiben und hoffe, dass die MDCG uns alle positiv überraschen wird, ganz nach dem Motto 'Kann der Hersteller reparieren, was er herstellt'.
Und auch die Hersteller müssen sich engagieren. Auch auf die Gefahr hin, wie eine kaputte Schallplatte zu klingen: Dies ist nicht der Zeitpunkt, um die Hände in den Schoß zu legen, Leute. Wie oben erläutert, sind die ersten Terminhürden für das MDR-QMS und die Einreichung eines Antrags auf Konformitätsbewertung bei einer benannten Stelle, die Sie möglicherweise noch finden müssen und die bereit sein muss, für Sie zu arbeiten und mit der Sie bis zum 26. Mai 2024 eine Konformitätsbewertungsvereinbarung unterzeichnen müssen, näher als Sie denken. Wenn Ihr Management glaubt, dass weniger als Vollgas in irgendeiner Weise eine gute Idee für den MDR-Übergang ist, ist es nicht für den Zweck geeignet. Der Vorschlag macht die MDR-Umstellung mehr denn je zu einer Chefsache, wie man auf Deutsch so schön sagt.
Annahmeverfahren
Der Verabschiedungsprozess folgt demselben Verfahren wie bei der überstürzten Verlängerung des Anwendungszeitraums der MDR im Jahr 2020: dem beschleunigten Mitentscheidungsverfahren, das seinerzeit in weniger als einem Monat formal blitzschnell abgeschlossen wurde. Dieser Vorschlag ist weitaus komplizierter, so dass ich davon ausgehe, dass sich die institutionellen Akteure etwas mehr Zeit nehmen werden (auch einige Parlamentarier haben angekündigt, dass sie sich nicht wie beim letzten Mal beeilen werden). Es ist möglich, dass es noch Änderungen am Text gibt.
Also bitte schön mit Zucker drauf: keine juristisch geschulten Berater, bitte nicht sagen, dass dieser Vorschlag es ist, dass er offiziell ist, dass er im Amtsblatt veröffentlicht wurde (was bis zur Verabschiedung noch nicht der Fall sein wird) - keine voreiligen Schlüsse ziehen, das Recht den Juristen überlassen und abwarten, bis das Gesetzgebungsverfahren seinen Lauf genommen hat. Erst dann werden wir wissen, wie das verabschiedete Gesetz aussehen wird und welche Regeln zu beachten sind.
Vom 11. bis 18. Januar findet eine Konsultation zu dem Vorschlag statt, der ursprünglich und offenbar irrtümlich für die normale Konsultationsfrist bis weit in den März hinein vorgesehen war, was von der Kommission eilig korrigiert wurde. Sie können immer noch Kommentare zu dem Vorschlag einreichen, aber erwarten Sie nicht, dass sie den Vorschlag tatsächlich ändern, da die Kommission einige der Kommentare an den Rat oder das Parlament weiterleiten kann (oder auch nicht). Wenn Sie die Dinge beeinflussen wollen, wenden Sie sich direkt an die Parlamentarier, die an diesem Dossier arbeiten, oder an die Mitgliedstaaten im Rat.
Zum jetzigen Zeitpunkt rechne ich selbst damit, dass der Vorschlag im März, wahrscheinlich in der zweiten Märzhälfte, angenommen wird und in Kraft tritt (unter Umgehung der normalen 20-tägigen Wartezeit nach der Veröffentlichung im Amtsblatt wegen der Eile).
Ausgleich zwischen den MDR- und IVDR-Fristzeiten
Interessanterweise wird die IVDR, wenn der Vorschlag angenommen wird, eine viel mildere und einfachere Regelung der Neuheitsschonfrist haben als die MDR - ist das sinnvoll, ist das fair? Beide haben nach Risikoklassen gestaffelte tilgungsfreie Zeiten und gingen von einer einzigen tilgungsfreien Zeit aus. Die IVDR hat jedoch fast keine Bedingungen für die Inanspruchnahme der tilgungsfreien Zeit, während die MDR sehr viele und offen gesagt komplexe Bedingungen hat.
Werden wir unter der IVDR ähnliche Bedingungen wie unter 120 (3d) sehen, wenn die tilgungsfreien Zeiten für die IVDR nicht ausreichen? Das könnte der Fall sein, denn wie wir gesehen haben, beeinflussen sich die MDR und die IVDR immer wieder gegenseitig bei den Lösungen, die für das Funktionieren der Übergangsregelung gewählt wurden. Ich würde die IVDR-Hersteller ermutigen, die derzeit verfügbaren Kapazitäten der benannten Stellen so gut wie möglich zu nutzen, um zu vermeiden, dass sie sich in Zukunft mit einer panischen Änderung der IVDR-Regelung auseinandersetzen müssen.
Kann der Hersteller reparieren, was er herstellt?
Wie oben erläutert, lässt der Vorschlag viele Fragen offen, z. B. die Frage, was ein Ersatzgerät ist. Offene Enden sind genau das, was wir zum jetzigen Zeitpunkt nicht brauchen, vor allem, weil sie eine Anleitung der MDCG erfordern, um sie zu erklären, und die MDCG-Anleitung ist nie schnell oder nach einem verlässlichen Zeitplan. Wenn möglich, sollten diese Punkte im Gesetzgebungsverfahren geregelt werden, weil wir sonst zur Klärung auf die MDCG-Leitlinien angewiesen sind, was einfach zu lange dauern wird.
Eine Sache, die uns Juristen Sorgen bereitet, ist die Frage der ex tunc Ungültigkeit von Zertifikaten oder der ex nunc Ungültigkeit bei Nichterfüllung der Bedingungen für die Verlängerung. Dies ist keine triviale Formalität und sollte zumindest in einem Erwägungsgrund des Vorschlags behandelt werden.
Erfreulich ist, dass zumindest einige der (großen) benannten Stellen der Meinung sind, dass der Vorschlag ihnen genügend Zeit lässt. Das ist in der Tat sehr positiv. Die Frage ist, ob dies auf alle benannten Stellen zutrifft, und was die Mitgliedstaaten tun werden, um sicherzustellen, dass die Maßnahmen des Positionspapiers zur MDCG 2022-14 von den benannten Stellen maximal genutzt werden können, und um zu überwachen, dass sich die benannten Stellen ernsthaft mit diesen Punkten befassen. An diesem Punkt sehe ich einige der kleineren benannten MDR-Stellen in ernsthaftem Durcheinander, auf Kosten ihrer armen Kunden, die in der Mitte gefangen sind, ohne angemessenen Rechtsbehelf und gefangen durch das Chaos, das die benannte Stelle anrichtet.
Die ursprüngliche Neuheitsschonfrist nach Artikel 120 (3) MDR war ausländischen Behörden in Ländern, die Wert auf die CE-Kennzeichnung legen, bereits schwer zu erklären und wurde oft weithin missverstanden, was zu Situationen führte, in denen sich ausländische Behörden schlichtweg weigerten, etwas anderes als eine glänzende neue MDR-Bescheinigung zu akzeptieren, weil das für sie Sinn machte. Die neue Neuheitsschonfrist wird ausländischen Behörden und den Behörden der Mitgliedstaaten, die Freiverkaufszertifikate ausstellen, noch schwerer zu erklären sein. Die Kommission und die Mitgliedstaaten müssen sich bemühen, dies international zu erklären, um zu vermeiden, dass überall Verwirrung entsteht. Das EU-Regulierungssystem für Medizinprodukte hat bereits zu viel von seinem internationalen Ruf eingebüßt, wir brauchen das nicht noch zu verschlimmern. Außerdem bleibt abzuwarten, wie ausländische Behörden mit der Abschaffung der Verkaufsfrist umgehen werden, denn diese für immer veralteten Produkte können rechtmäßig auf dem Unionsmarkt verkauft werden, was bedeutet, dass sie durch eine Freiverkaufsbescheinigung abgedeckt werden können. Das bedeutet, dass wir den ausländischen Behörden möglicherweise noch lange Zeit erklären müssen, wie das funktioniert.
Wie ich bereits gesagt habe: Entscheidend für den Erfolg des Vorschlags ist die proaktive Einführung des MDCG 2022-14 und ein funktionierendes Sicherheitsnetz ab dem MDCG 2022-18. Dies liegt vollständig in den Händen der MDCG und der Mitgliedstaaten, die jetzt mehr denn je die Gelegenheit haben, zu zeigen, dass die Medizinproduktepolitik eine ernste Angelegenheit ist, und so viel Teil der Lösung zu sein, wie sie können.
Aber erst einmal wollen wir sehen, wie sich das Gesetzgebungsverfahren entwickelt und wie der Vorschlag aussieht, wenn er angenommen wird.























